


Cancer cells have a high demand for biomass—cellular components such as proteins, nucleic acids, and lipids—to support their rapid growth and division. One way in which they attain biomass in the nutrient-poor regions found throughout the tumor microenvironment (TME) is by deregulating or reprogramming cellular metabolism pathways. This allows tumors to generate energy, biosynthetic intermediates, and reducing equivalents (e.g., NADH, NADPH, and FADH2) necessary for continuous cell division, as well as to evade immune attack by sequestering nutrients away from effector T cells.1,2
This blog explores some of the major biochemical and signaling pathways associated with Deregulating Cellular Metabolism, a next-generation Hallmark of Cancer, and highlights key proteins and research targets being studied for therapeutic intervention.
< Jump to the product list at the end of this blog >
Biochemical and Signaling Pathways in Cancer Metabolism
Hypoxia Signaling and HIF-1
|
Oxygen (O2) is essential for aerobic life, and cells have evolved sophisticated molecular machinery to sense and respond to oxygen levels. A key player in this response is Hypoxia-Inducible Factor-1 (HIF-1), a transcription factor composed of an oxygen-regulated α-subunit and a constitutively expressed β-subunit. Under well-oxygenated (normoxic) conditions, HIF-1α is hydroxylated by oxygen-dependent prolyl 4-hydroxylase domain (PHD) proteins. Hydroxylated HIF-1α then binds to the von Hippel–Lindau (VHL) tumor suppressor, leading to its rapid degradation.2 |
|
What are the Hallmarks of Cancer?The Hallmarks of Cancer1-3 are a research framework that organizes the key traits cancer cells acquire in order to grow and spread. Initially described by Douglas Hanahan and Robert Weinberg in 2000, the framework groups the underlying mechanisms of cancer into a series of smaller subsets to advance discovery. The concept was expanded in 2011 to include two additional hallmarks and two enabling characteristics, and again in 2022 with four new emerging hallmarks.
|
 Deregulating Cellular Metabolism is one of the next-generation Hallmarks of Cancer, identified in 2011.
Deregulating Cellular Metabolism is one of the next-generation Hallmarks of Cancer, identified in 2011.However, under hypoxic (low oxygen) conditions, which are common in the tumor microenvironment (TME), non-hydroxylated HIF-1α accumulates, associates with HIF-1β to activate the HIF-1 complex, and translocates to the nucleus. The HIF-1 complex then activates the transcription of target genes such as VEGF, EPO, Glut1, as well as genes for various glycolytic enzymes.2
Western blot (WB) analysis of extracts from three cell lines: Hep G2 (human liver cancer cells), Raji cells (human Burkitt lymphoma B cells), and U-2 OS cells (human osteosarcoma cells). Each cell line was either untreated (-), treated with cobalt chloride (Hep G2 and Raji), or treated with DMOG (U-2 OS), and analyzed using HIF-1 alpha (D1S7W) Rabbit Monoclonal Antibody #36169 (upper) or beta-Actin (D6A8) Rabbit Monoclonal Antibody #8457 (lower). Cobalt chloride and DMOG interfere with HIF-1α degradation, leading to its accumulation and mimicking the effects of hypoxia.
This shifts cells toward hypoxic metabolism and increases processes such as blood vessel formation (angiogenesis) and glucose uptake, helping cancer cells to survive and proliferate despite the low oxygen levels of the TME.2,4
 |
Download the Hypoxia Signaling Pathway Diagram to explore key protein targets and associated CST products. |
Blog: Hypoxia & Cancer: The role of HIF-1α in Oxygen Sensing, Metabolism, & Tumorigenesis
Glucose Metabolism and the Warburg Effect
Glucose is the main fuel source for most cells. Its uptake from the bloodstream is mediated by glucose transporters such as Glut1 (SLC2A1), which is regulated via the PI3K/Akt pathway. In many types of cancer, the activation of this pathway—and the expression of Glut1—is upregulated to support the increased transport of glucose into cancer cells and fuel their high metabolic demands.2,4 After transport into the cell, glucose is metabolized by glycolysis to produce pyruvate, which serves as a crucial link to other metabolic pathways.

Immunohistochemical (IHC) analysis of paraffin-embedded human renal cell carcinoma using Glut1 (E4S6I) Rabbit Monoclonal Antibody #73015, showing expression of the glucose transporter Glut1, which is frequently elevated in tumor cells to support increased glucose metabolism.
In normal resting cells with sufficient oxygen, pyruvate is transported from the cytoplasm into the mitochondria, where it enters the tricarboxylic acid (TCA) or Krebs cycle as part of aerobic respiration to efficiently generate energy in the form of ATP. Under hypoxia, lactate dehydrogenase (LDHA) converts pyruvate to lactate, allowing NADH to be oxidized back to NAD+, and enabling glycolysis to continue.
Cancer cells, however, convert large amounts of glucose to lactate, even under normal oxygen levels—a phenomenon known as aerobic glycolysis or the Warburg effect. The Warburg effect generates increased levels of ATP, biosynthetic intermediates, and reducing equivalents to fuel cancer cell proliferation.2,4
 |
Download the Warburg Effect Pathway Diagram to explore key protein targets and associated CST products. |
A key metabolic checkpoint between glycolysis and the TCA cycle is controlled by pyruvate dehydrogenase (PDH) α1 (PDHA1). In many cancer cells, PDHA1 is inactivated by phosphorylation at Ser293, a modification mediated by pyruvate dehydrogenase kinase 1 (PDHK1). This phosphorylation inhibits PDHA1 activity, blocking the decarboxylation of pyruvate to acetyl-CoA and therefore inhibiting the TCA cycle and pushing cells toward lactate production in the presence of oxygen.4,5

Flow cytometric analysis of HeLa cells, untreated (green) or λ phosphatase-treated (blue) using Phospho-Pyruvate Dehydrogenase alpha1 (Ser293) (E4V9L) Rabbit Monoclonal Antibody #37115 (solid lines) or concentration-matched Rabbit (DA1E) Monoclonal Antibody IgG Isotype Control #3900 (dashed lines). The phosphorylation of PDHA1 at Ser293 inactivates the enzyme and inhibits pyruvate from entering the TCA cycle, a modification associated with the Warburg effect. The measured reduction in phospho-PDHA1 (Ser293) levels after λ phosphatase treatment helps confirm antibody specificity.
Other critical enzymes include PKM2—which catalyzes the final rate-limiting step of glycolysis—and LDHA, which converts pyruvate to lactate. PKM2 ensures high glycolytic throughput, while LDHA maintains the NAD+/NADH balance required by glycolysis. Both enzymes are critical for the Warburg effect and are frequently upregulated in cancer cells.
Blog: Interrogating Immunometabolism in the TME Using a Metabolic Checkpoint
Glutamine Dependence in Tumors
Glutamine is another critical fuel source for cancer metabolism. It supplies both the carbon necessary for the TCA cycle, as well as the γ-nitrogen (amide nitrogen) required for the creation of purine and pyrimidine nucleotides, which are vital for the DNA and RNA production in rapidly dividing cancer cells. Glutamine is also required for the synthesis of asparagine, an amino acid that activates mTORC1, a major kinase complex of the mTOR signaling pathway. This pathway also promotes the biosynthesis of purines and pyrimidines, further supporting nucleotide production.2,4
Glutamine enters cells through the amino acid transporter ASCT2 (SLC1A5) and is converted to glutamate in the mitochondria by glutaminase (GLS). It is further processed by glutamate dehydrogenase (GDH) to form the TCA cycle intermediate α-ketoglutarate (α-KG). The process of replenishing TCA cycle intermediates from precursors that are not TCA cycle intermediates themselves is called anaplerosis. Glutamine also plays a role in maintaining redox homeostasis via the antioxidant glutathione (GSH) and the generation of NADPH.5


Many cancer cells demonstrate high levels of glutamine uptake and utilization, but the signaling involved in this process is not fully understood, making this pathway a key focus area for cancer researchers. Specifically, the oncogene c-Myc has been highlighted in recent studies and has been shown to drive metabolic reprogramming by upregulating the expression of genes involved in glutamine transport (ASCT2) and metabolism (GLS1).2,4,5

Lipid Metabolism in Cancer
Lipids serve as membrane components, energy storage, and signaling molecules. When limited glucose is available, cancer cells use lipids as an alternative energy source. The expression of lipid transport proteins, such as CD36, FATP1, FATP2, and FABP4, is frequently upregulated in cancer cells. Cancer cells are also known to increase lipid biosynthesis in order to generate new plasma membranes required for cell proliferation.4
Proteins with key roles in harnessing lipid metabolism for cancer progression include SREBP-1, a master transcriptional regulator, and its downstream targets, such as Fatty Acid Synthase (FASN) and ATP-Citrate Lyase (ACLY). ACLY is essential for converting citrate into acetyl-CoA, a precursor for fatty acid and cholesterol biosynthesis that is often upregulated in cancer cells to support increased membrane production and metabolic demands.


In addition, CPT1A is a critical enzyme that catalyzes the rate-limiting step in fatty acid beta-oxidation by facilitating the transport of long-chain fatty acids into the mitochondria, a process that is upregulated by cancer cells to support their energy needs and promote tumor development.4
Autophagy & Tumor Nutrient Acquisition
In addition to the metabolic signaling pathways described above, cancer cells can also manipulate autophagy to promote tumor growth and survival under different conditions. Autophagy is a lysosome-dependent cellular degradation pathway that, under normal conditions, helps remove unnecessary and damaged cellular components, reduces stress, and maintains cellular homeostasis. The relationship between autophagy and cancer is incredibly complex, and cancer cells can benefit from both its promotion and inhibition.6-8

In tumors, increased autophagy can promote tumor growth by preventing apoptosis and breaking down cellular components to release nutrients. These nutrients, which include amino acids, nucleotides, and fatty acids, are often limited in the tumor microenvironment (TME), and are used to enable increased cellular metabolism in cancer cells. In addition, tumor-intrinsic autophagy enhances immune evasion in some cancers by blocking antigen presentation and immune-mediated tumor targeting.
Key autophagy-associated proteins are often upregulated or dysregulated in cancer. For example, LC3B is a widely used marker of autophagosome formation,6,7 while SQSTM1/p62 is an autophagy marker that is important for cancer initiation and progression.8 TFEB is a master regulator of autophagy and lysosomal biogenesis and can function as an oncogene by upregulating lysosomal and metabolic pathways.9-12

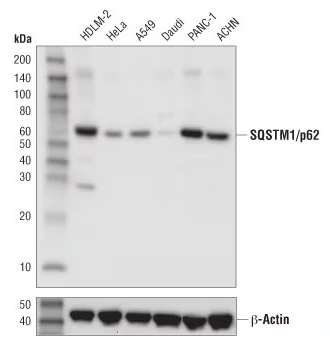
However, autophagy alone cannot supply cells with all the biomass needed for their proliferation. Cancer cells also use opportunistic modes of nutrient acquisition, such as macropinocytosis, to acquire new biomass. This involves the extension of membrane protrusions into the extracellular environment to engulf amino acids and other macromolecules in structures known as macropinosomes, which are then trafficked to lysosomes for degradation. Genetic alterations in cancer cells, such as Ras mutations, can increase the rate and volume at which macropinocytosis occurs, helping to support tumor growth under nutrient-deficient conditions.4
In addition to its role in tumor promotion, autophagy can also act as a tumor suppressor under certain conditions. Through mitophagy, the controlled removal of damaged mitochondria, autophagy lowers the release of reactive oxygen species (ROS) and limits excess glycolytic activity, both of which can drive cancer progression. Despite the Warburg effect’s reduced demand for mitochondrial ATP, cancer cells still rely on mitochondria to generate the intermediaries needed for metabolic pathways. Mitophagy is activated in response to various cellular stresses, such as hypoxia, nutrient deprivation, and inflammation. When mitophagy signaling is disrupted, damaged mitochondria can accumulate, leading to impaired nutrient and energy homeostasis, increased oxidative stress, and an increased susceptibility to cell death.9
Therapeutic Opportunities Targeting Cancer Metabolism
Metabolic reprogramming in cancer cells offers a wealth of opportunities for therapeutic intervention. Many of the pathways discussed above are being investigated for therapeutic targeting, including through the inhibition of glutaminases and lipogenic enzymes.2,4,5
Another promising therapeutic strategy involves blocking enzymes involved in nucleotide synthesis, such as ribonucleotide reductase (RNR) and dihydroorotate dehydrogenase (DHODH), to disrupt the production of deoxyribonucleotides and ultimately halt cancer cell proliferation.
A key challenge for cancer research lies in understanding the varied metabolic adaptations in different types of cancer cells, as well as addressing ever-evolving metabolic changes in the same type of cancer cells during cancer progression. Continued investigation is essential to develop effective treatments in cancer metabolism.2,4,5
Explore select relevant CST antibody products for studying cancer metabolism using the table below.
Additional Resources
Read the additional blog posts in the Hallmarks of Cancer series to learn more:
- Evading Growth Suppressors
- Nonmutational Epigenetic Reprogramming
- Avoiding Immune Destruction
- Tumor-Promoting Inflammation
- Activating Invasion & Metastasis
- Inducing or Accessing Vasculature (Angiogenesis)
- Senescent Cells
- Genome Instability & Mutation
- Resisting Cell Death
- Unlocking Phenotypic Plasticity
- Sustaining Proliferative Signaling
Select References
- Hanahan D, Weinberg RA (January 2000). "The Hallmarks of Cancer". Cell. 100 (1): 57–70. doi:10.1016/S0092-8674(00)81683-9
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-674. doi:10.1016/j.cell.2011.02.013.
- Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discov. 2022;12(1):31-46. doi:10.1158/2159-8290.CD-21-1059.
- Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016;23(1):27-47. doi:10.1016/j.cmet.2015.12.006
- Brisimi TS, Christou CD, Katsiampoura A, et al. Targeting cancer metabolism—new therapeutic opportunities for cancer therapy. Cancers (Basel). 2023;15(14):3725. doi:10.3390/cancers15143725.
- Fontana R, Foti M, Gouttenoire J. Immunostaining of autophagic markers in cancer: LC3B, p62, and beyond. Clin Cancer Res. 2012;18(2):370-379. doi:10.1158/1078-0432.CCR-11-1282.
- Rangwala R, Chang YC, Hu J, et al. Combined MTOR and autophagy inhibition: a phase 1 trial with temsirolimus and hydroxychloroquine in patients with advanced solid tumors and melanoma. Autophagy. 2013;9(5):898-899. doi:10.4161/auto.23371.
- Hu YB, Shen N, Wang XB, et al. SQSTM1/p62 in autophagy-related immune responses and cancer. Front Immunol. 2022;13:852298. doi:10.3389/fimmu.2022.852298.
- Settembre C, Fraldi A, Medina DL, Ballabio A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol. 2013;14(5):283-296. doi:10.1038/nrm3565.
- Medina DL, Fraldi A, Bouche V, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332(6036):1429-1433. doi:10.1126/science.1204592.
- Li L, Li S, Cai H. Recent progress in the research of TFEB in cancer. Cancer Manag Res. 2023;15:1619-1629. doi:10.2147/CMAR.S369876.
- Li Y, Zhu X, Zeng Y, et al. TFEB promotes hepatocellular carcinoma progression by upregulating PD-L1 via binding to the IRF1 promoter. Cell Death Dis. 2023;14(3):187. doi:10.1038/s41419-023-05667-8.


/Binary_oasis_Blog%20Header.webp)
