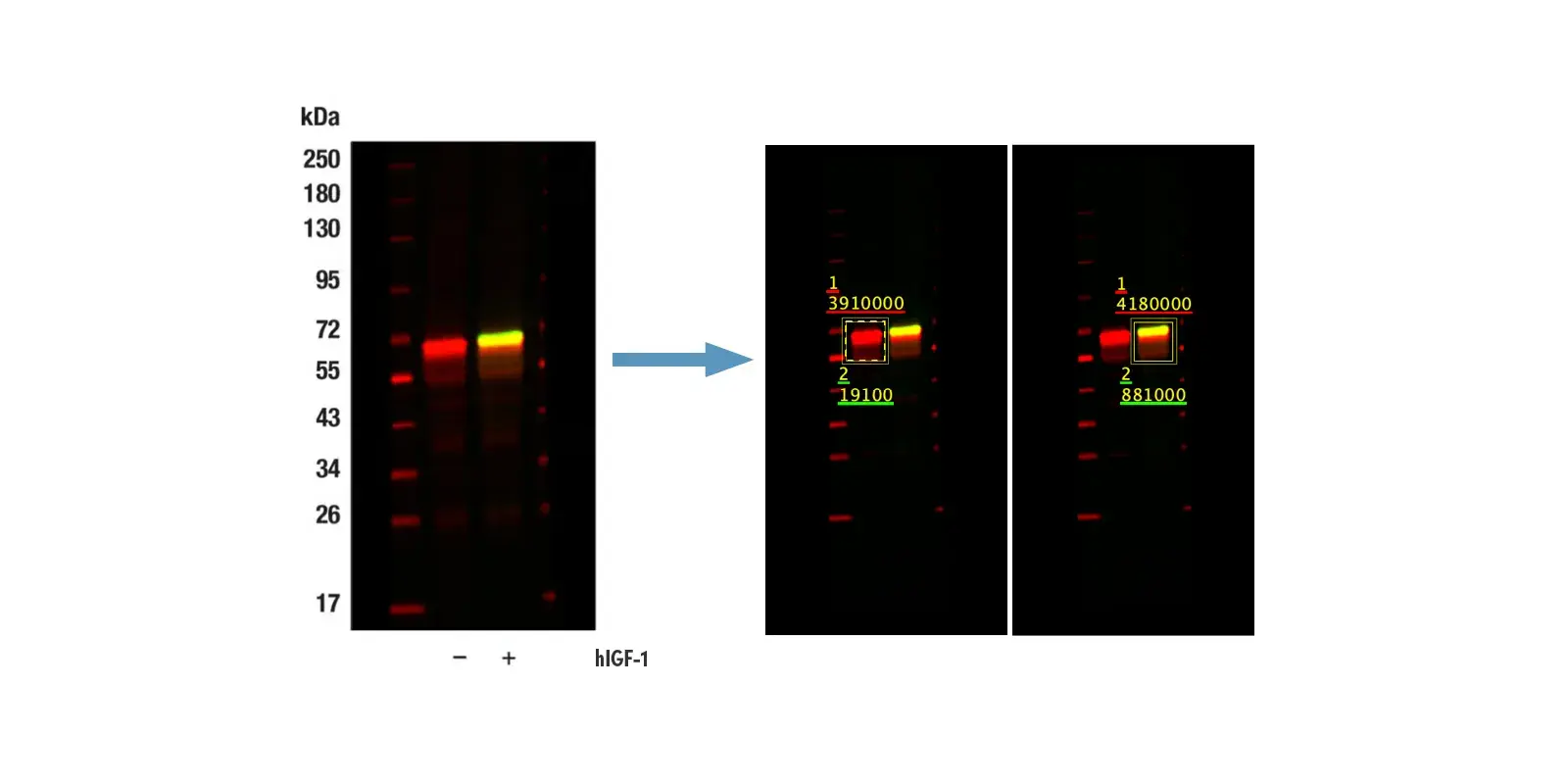
One of the defining characteristics of cancer cells is their ability to avoid programmed death—especially apoptosis. Under normal conditions, apoptosis plays a critical role in maintaining tissue homeostasis and eliminating damaged or unwanted cells. In cancer cells, however, disruptions or imbalances in apoptotic machinery allow cells to survive DNA damage and oncogenic signaling that would typically trigger death—instead favoring uncontrolled proliferation.
This blog focuses on the key protein families that regulate apoptosis, and touches on alternative cell death pathways—such as necroptosis, ferroptosis, and pyroptosis—that are being targeted for therapeutic intervention within the Hallmark of Cancer Resisting Cell Death.
< Jump to the product list at the end of this blog >
|
Overview of Apoptosis Apoptosis is a highly regulated form of cell death initiated either through external signals (the extrinsic pathway) or internal stress (the intrinsic, mitochondrial pathway). Regardless of how it’s initiated, both apoptotic pathways ultimately lead to the activation of caspases, cysteine proteases that play a crucial role in executing programmed cell death. |
What are the Hallmarks of Cancer?The Hallmarks of Cancer1-3 are a research framework that organizes the key traits cancer cells acquire in order to grow and spread. Initially described by Douglas Hanahan and Robert Weinberg in 2000, the framework groups the underlying mechanisms of cancer into a series of smaller subsets to advance discovery. The concept was expanded in 2011 to include two additional hallmarks and two enabling characteristics, and again in 2022 with four new emerging hallmarks.
|
 One of the original hallmarks, Resisting Cell Death was initially referred to as "Evading Apoptosis."
One of the original hallmarks, Resisting Cell Death was initially referred to as "Evading Apoptosis."The extrinsic pathway is typically initiated by caspase-8 activation, whereas the intrinsic pathway is initiated by the activation of caspase-9.
 Western blot (WB) analysis of extracts from HCT 116 and CRISPR/Cas9 caspase-8 knockout HCT 116 cells, untreated (-) or treated with Staurosporine #9953, using Cleaved Caspase-8 (Asp374) (E6H8S) Rabbit Monoclonal Antibody #98134 (upper), Caspase-8 (1C12) Mouse Monoclonal Antibody #9746 (middle), or GAPDH (D16H11) Rabbit Monoclonal Antibody #5174 (lower). Staurosporine treatment triggers apoptosis, leading to the activation of caspase-8.
Western blot (WB) analysis of extracts from HCT 116 and CRISPR/Cas9 caspase-8 knockout HCT 116 cells, untreated (-) or treated with Staurosporine #9953, using Cleaved Caspase-8 (Asp374) (E6H8S) Rabbit Monoclonal Antibody #98134 (upper), Caspase-8 (1C12) Mouse Monoclonal Antibody #9746 (middle), or GAPDH (D16H11) Rabbit Monoclonal Antibody #5174 (lower). Staurosporine treatment triggers apoptosis, leading to the activation of caspase-8.
Importantly, aberrant regulation of extrinsic apoptosis through the interaction of Fas ligand (FasL) with its receptor, Fas, plays a key role in tumor immune evasion and metastasis, as cancer cells can evade death receptor-mediated signals that otherwise support immune surveillance.

Once activated, both pathways trigger a cascade of additional caspases, including the executioner caspases, such as caspase-3. This results in the cleavage of several proteins, including poly (ADP-ribose) polymerase (PARP), lamins, and DNA fragmentation factor 45 (DFF45), which are involved in cell disassembly.


How Cancer Cells Outsmart Apoptosis
Cancer cells alter protein expression to bypass cell death signals, allowing them to persist despite damage or stress that would normally eliminate abnormal cells. These adaptations include upregulating anti-apoptotic proteins, silencing pro-apoptotic genes, and interfering with the activation of death pathways.
Modulating Expression of the IAP Protein Family
The Inhibitor of Apoptosis Protein (IAP) family is a group of apoptosis inhibitors that suppresses programmed cell death by inhibiting caspases. Key family members, including cellular inhibitor of apoptosis 1 (c-IAP1), cellular inhibitor of apoptosis 2 (c-IAP2), X-linked inhibitor of apoptosis protein (XIAP), survivin, and livin, suppress apoptosis by directly binding to caspases through baculovirus inhibitor repeat (BIR) domains.


IAPs are frequently overexpressed in tumors, resulting in resistance to apoptosis. The inhibition of IAPs by antagonists such as Smac/Diablo, which is released from the mitochondria during apoptosis, also disrupts IAP-caspase interactions and promote cell death. They may also be induced in cancer by pro-survival pathways such as NF-kB signaling.
Disrupting Pro- and Anti-Apoptotic Signals via the Bcl-2 Family
A cell’s sensitivity to intrinsic apoptosis is controlled by levels of pro- and anti-apoptotic members of the B-cell lymphoma 2 (Bcl-2) family. Specifically, Bcl-2, along with Bcl-xL, Bcl-w, Mcl-1, and A1/Bfl-1, are anti-apoptotic family members that bind to and inhibit the pro-apoptotic family members Bax, Bak, and Bok. When not inhibited, Bax and Bak dimerization causes mitochondrial outer membrane permeabilization (MOMP) and the release of apoptotic-inducing proteins from the mitochondria, including cytochrome c and Apoptosis-Inducing Factor (AIF). Once in the cytoplasm, cytochrome c associates with Apaf-1 to trigger the activation of caspase-9.
 IHC analysis of paraffin-embedded human lung carcinoma, using Bcl-xL (54H6) Rabbit Monoclonal Antibody #2764.
IHC analysis of paraffin-embedded human lung carcinoma, using Bcl-xL (54H6) Rabbit Monoclonal Antibody #2764.
Resistance to cell death in cancer cells is often the result of elevated levels of anti-apoptotic Bcl-2 family members, which promote cellular survival. For example, Bcl-2 was identified as a proto-oncogene overexpressed as a result of chromosomal translocation in follicular lymphomas.
BH3-Only Proteins: The Gatekeepers of Apoptosis
The regulation of apoptosis by the Bcl-2 family is facilitated by the BH3 binding domain, which is shared between the pro- and anti-apoptotic proteins, as well as a third group called the “BH3-only” proteins. BH3-only proteins, including Bid, Bim, Bad, BMF, Puma, Noxa, and Hrk, can interact with and neutralize anti-apoptotic members (e.g., Bcl-2, Bcl-xL) or directly activate effectors Bax and Bak, facilitating mitochondrial outer membrane permeabilization (MOMP) and cell death.
Cells have evolved multiple mechanisms for regulating BH3-only proteins, including phosphorylation, cleavage, and transcriptional regulation—many of which are targeted by cancer cells to promote disease progression. A classic example of BH3-only protein regulation includes the caspase-induced cleavage of Bid, which promotes its death-inducing function.
 IHC analysis of paraffin-embedded human colon adenocarcinoma using Bim (C34C5) Rabbit Monoclonal Antibody #2933 performed on the Leica BOND Rx. Bim is a pro-apoptotic protein belonging to the BH3-only subgroup of Bcl-2 family members.
IHC analysis of paraffin-embedded human colon adenocarcinoma using Bim (C34C5) Rabbit Monoclonal Antibody #2933 performed on the Leica BOND Rx. Bim is a pro-apoptotic protein belonging to the BH3-only subgroup of Bcl-2 family members.
Other examples include Akt-mediated phosphorylation of Bad, which impairs its ability to induce apoptosis. Conversely, the loss of tumor suppressor p53, a transcription factor that regulates cell cycle arrest and apoptosis, impairs the transcription of Puma and Noxa, thereby reducing apoptosis.
Current Therapeutic Directions
Targeting Apoptosis in Cancer Cells
Therapeutic strategies targeting apoptotic signaling have led to major advances in cancer treatment, and current research focuses on targeting the interaction domains within IAP and Bcl-2 protein families to overcome resistance to apoptosis. For example, the Bcl-2 inhibitor venetoclax is approved by the US Food and Drug Administration for treating B-cell lymphomas with elevated levels of Bcl-2, such as those with Bcl-2 gene translocations. Smac mimetics, which inhibit IAPs, have been tested in many clinical trials, as have BH3 mimetics, which block anti-apoptotic proteins such as Bcl-2, Bcl-xL, and Mcl-1. Broader-spectrum BH3 mimetics—for example, the Bcl-2/Bcl-xL/Bcl-w inhibitor navitoclax—and emerging Mcl-1 inhibitors have also shown therapeutic promise, though clinical trials have been limited by on-target toxicities.
 WB analysis of extracts from U-118 MG and Detroit 562 cells, untreated (-) or treated with SMAC mimetic SM-164 #56003 (100nM, 6 hr; +), using c-IAP1 (F7C3U) Rabbit Monoclonal Antibody #96099 (upper) or GAPDH (D16H11) Rabbit Monoclonal Antibody #5174 (lower). SM-164 is a SMAC mimetic that reduces c-IAP1 expression and may help overcome tumor cell resistance to apoptosis.
WB analysis of extracts from U-118 MG and Detroit 562 cells, untreated (-) or treated with SMAC mimetic SM-164 #56003 (100nM, 6 hr; +), using c-IAP1 (F7C3U) Rabbit Monoclonal Antibody #96099 (upper) or GAPDH (D16H11) Rabbit Monoclonal Antibody #5174 (lower). SM-164 is a SMAC mimetic that reduces c-IAP1 expression and may help overcome tumor cell resistance to apoptosis.
These approaches remain an active research area as efforts continue to expand the therapeutic window, potentially through improved PROTAC design or combination therapy with conventional chemotherapeutic agents.
Beyond Apoptosis: Reactivating Ferroptosis, Necroptosis & Pyroptosis Pathways
Beyond apoptosis, recent studies are also exploring the role of non-apoptotic cell death pathways in cancer. Many tumor types have evolved mechanisms to evade these forms of regulated cell death, making them attractive targets for new therapeutic strategies. Areas of focus include regulated necrotic pathways such as ferroptosis, necroptosis, and pyroptosis.

Importantly, these non-apoptotic pathways can both restrain and promote cancer—while their activation can suppress tumor growth by providing alternative ways to kill cancer cells resistant to apoptosis, the associated activation of inflammation and immune response within the tumor microenvironment can, in some contexts, facilitate tumor progression, metastasis, or immune evasion.
Ferroptosis
Ferroptosis is an iron-dependent form of programmed cell death characterized by the accumulation of lipid peroxides in cell membranes, leading to loss of membrane integrity and cell death. A central regulator of this pathway is the antioxidant enzyme GPX4, which detoxifies lipid peroxides but depends on glutathione. In turn, glutathione is synthesized from cysteine derived from cystine imported by the transporter xCT/SLC7A11.
Under normal conditions, the tumor suppressor p53 limits xCT/SLC7A11 expression, but when p53 is mutated or lost—as in many cancers—xCT/SLC7A11 is upregulated, increasing cystine uptake, sustaining GPX4 activity, and preventing ferroptosis. Therapeutically, ferroptosis can be modulated at multiple nodes, including GPX4, xCT/SLC7A11, iron handling, cellular antioxidant systems, and lipid-metabolizing enzymes such as ACSL4.

WB analysis of extracts from various cell lines using ACSL4 (F6T3Z) Rabbit Monoclonal Antibody #38493 (upper) or GAPDH (D16H11) Rabbit Monoclonal Antibody #5174 (lower). Low expression of ACSL4 protein in MCF7 and T-47D cells is consistent with the predicted expression pattern.
Inducing ferroptosis may be useful for killing tumor cells, yet the immunosuppressive and pro-tumorigenic effects of ferroptotic cell death in some contexts also support the development of ferroptosis inhibitors, underscoring that both activation and inhibition of this pathway may have clinical value.
Necroptosis
Necroptosis is a lytic, pro-inflammatory form of programmed cell death that involves the rupture of cell membranes and the release of cellular contents, triggering an immune response. In response to inflammatory cytokines such as TNFα, RIPK1 is recruited to receptor complexes and, when caspase‑8–mediated apoptosis is blocked, its kinase activity promotes activation of RIPK3, which then phosphorylates MLKL to execute necroptosis. In many tumors, RIPK3 is epigenetically silenced via promoter methylation, which disables necroptosis. Experimental approaches include strategies to modulate RIPK1 and related signaling in combination with immune checkpoint inhibitors to enhance antitumor immunity.
 IF analysis of L-929 cells, untreated (left), pre-treated with Z-VAD, followed by treatment with SM-164 and mTNF-α (center), and then post-processed with λ-phosphatase (right), using Phospho-MLKL (Ser345) (D6E3G) Rabbit Monoclonal Antibody #37333 (green). Red = Propidium Iodide (PI)/RNase Staining Solution #4087 (fluorescent DNA dye).
IF analysis of L-929 cells, untreated (left), pre-treated with Z-VAD, followed by treatment with SM-164 and mTNF-α (center), and then post-processed with λ-phosphatase (right), using Phospho-MLKL (Ser345) (D6E3G) Rabbit Monoclonal Antibody #37333 (green). Red = Propidium Iodide (PI)/RNase Staining Solution #4087 (fluorescent DNA dye).
Pyroptosis
Pyroptosis is a pro-inflammatory form of programmed cell death mediated by gasdermins. In this pathway, the activation of inflammatory caspases (such as caspase-1) leads to the cleavage of Gasdermin D (GSDMD), which forms pores in the cell membrane and results in cell swelling, lysis, and release of inflammatory cytokines. In cancer, genes responsible for pyroptosis—such as gasdermin E (GSDME)—are often epigenetically silenced, reducing the cell’s ability to undergo inflammatory death and evade immune detection. Experimental strategies to restore or modulate pyroptosis include agents that activate pyroptotic caspases or upregulate gasdermin expression, but further studies are needed to determine the contexts in which pathway activation or inhibition will provide the greatest therapeutic benefit.
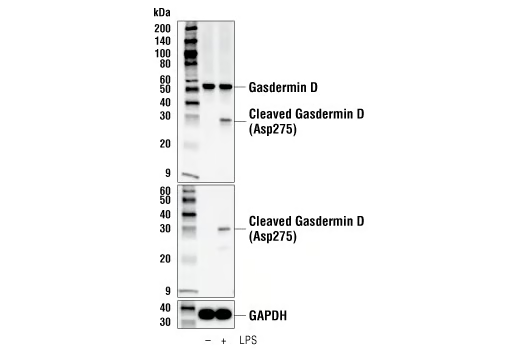 WB analysis of extracts from THP-1 cells, differentiated with TPA (12-O-Tetradecanoylphorbol-13-Acetate) #4174 (50 ng/ml, overnight) and then treated with Lipopolysaccharides (LPS) #14011 (5 μg/ml, 6 hr), using Gasdermin D (E9S1X) Rabbit Monoclonal Antibody #39754 (upper), Cleaved Gasdermin D (Asp275) (E7H9G) Rabbit Monoclonal Antibody #36425 (middle), or GAPDH (D16H11) Rabbit Monoclonal Antibody #5174 (lower).
WB analysis of extracts from THP-1 cells, differentiated with TPA (12-O-Tetradecanoylphorbol-13-Acetate) #4174 (50 ng/ml, overnight) and then treated with Lipopolysaccharides (LPS) #14011 (5 μg/ml, 6 hr), using Gasdermin D (E9S1X) Rabbit Monoclonal Antibody #39754 (upper), Cleaved Gasdermin D (Asp275) (E7H9G) Rabbit Monoclonal Antibody #36425 (middle), or GAPDH (D16H11) Rabbit Monoclonal Antibody #5174 (lower).
Redirecting Cell Death Pathways to Defeat Tumors
The ability of cancer cells to evade multiple forms of programmed cell death—including apoptosis, necroptosis, ferroptosis, and pyroptosis—is a major contributor to tumor progression and therapy resistance. Targeting these pathways to either promote tumor cell death or assist tumor-fighting immune cells in the tumor microenvironment holds great promise for cancer therapy. Advances in the development of small molecules, biologics, and combination strategies to restore or trigger regulated cell death are paving the way for more effective, long-lasting cancer treatments.
Sampler Kits & Monoclonal Antibodies for Studying Cell Death Pathways in Cancer
Additional Resources
- Learn about the key proteins and signaling events involved in intrinsic and extrinsic cell death in the blog Mechanisms of Cell Death: Apoptosis
- Read the additional blog posts in the Hallmarks of Cancer series to learn more:
- Evading Growth Suppressors
- Nonmutational Epigenetic Reprogramming
- Avoiding Immune Destruction
- Tumor-Promoting Inflammation
- Activating Invasion & Metastasis
- Inducing or Accessing Vasculature (Angiogenesis)
- Senescent Cells
- Genome Instability & Mutation
- Deregulating Cellular Metabolism
- Unlocking Phenotypic Plasticity
- Sustaining Proliferative Signaling
Select References:
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-674. doi:10.1016/j.cell.2011.02.013
- Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35(4):495-516. doi:10.1080/01926230701320337
- Liu ZL, Chen HH, Zheng LL, Sun LP, Shi L. Angiogenic signaling pathways and anti-angiogenic therapy for cancer. Signal Transduct Target Ther. 2023;8(1):198. Published 2023 May 11. doi:10.1038/s41392-023-01460-1
- Liu X, Zhang J, Yi T, et al. Decoding tumor angiogenesis: pathways, mechanisms, and future directions in anti-cancer strategies. Biomark Res. 2025;13(1):62. Published 2025 Apr 18. doi:10.1186/s40364-025-00779-x