Genetic alterations are a fundamental characteristic of cancer. When DNA mutations and chromosomal rearrangements affect the pathways that control how cells grow, divide, and repair their DNA, the regulatory mechanisms that prevent abnormal cellular behavior can break down. Known as genomic instability, mutations in these critical pathways predispose cells to the accumulation of genetic errors, which can be harnessed by cancer cells to drive tumor creation and progression.
This blog explores Genome Instability and Mutation, part of the Hallmarks of Cancer framework, and looks at some of the key proteins and signaling pathways being investigated for therapeutic targeting.
< Jump to the product list at the end of this blog >
How do genomic instability and mutations contribute to cancer?
DNA can become damaged in many ways, from internal errors like interference with DNA replication, cellular metabolism, and oxidative stress, to external sources like UV light, ionizing radiation, and chemical exposure.
 Genome Instability and Mutation was first described in 2011 as an enabling characteristic
Genome Instability and Mutation was first described in 2011 as an enabling characteristicHowever, if DDR mechanisms become defective or are overwhelmed, mutations and chromosomal errors can go unrepaired, allowing damaged DNA to persist. As errors accumulate, a cell’s genetic material becomes unstable, increasing the likelihood of mutations in critical genes that control cell cycle, cell proliferation, and programmed cell death.
These changes can give rise to traits that allow abnormal cells to survive and evolve into cancer.
Relevant Pathway Diagrams
 |
DNA Damage Response (DDR) Pathway: Explore the different types of DNA damage—including replication errors, crosslinks, and strand breaks—and the key repair pathways and proteins involved in detecting and fixing errors to maintain genome stability. |
 |
G1/S Checkpoint Pathway: Visualize the signals and proteins that regulate the decision for a cell to begin DNA replication and enter S phase, including growth factors, DNA damage sensors, and cell cycle regulators. |
 |
G2M/DNA Damage Checkpoint Pathway: See how cells monitor and respond to DNA damage before mitosis and the key proteins and signaling cascades that can delay cell division until genetic errors are repaired, helping preserve genome integrity by preventing entry into M phase. |
Types of DNA Repair
When DNA damage occurs, cells employ a variety of repair mechanisms to identify and correct different types of DNA lesions. The initial phase of the DDR is accomplished through DNA damage sensors (DDS), which detect faulty DNA structures using highly specific protein-DNA interactions.

Once bound to a DNA lesion, DDS trigger signaling cascades that activate DNA repair mechanisms, which include the following:
- Homologous Recombination (HR): A crucial pathway for repairing double-strand breaks (DSBs), homologous recombination uses undamaged, homologous DNA—most often on a sister chromatid—as a template to repair the chromosome with the damaged DNA. During HR, the MRN complex (Mre11, Rad50, and p95/NBS1) binds to a DSB, and together with CtIP, processes the DNA and activates the ATM checkpoint kinase (see section below). Rad51 then binds to the resulting single-stranded DNA to mediate repair, helped by auxiliary proteins Rad54 and BRCA2, which are recruited by BRCA1 and PALB2. The process is highly active during the S and G2 phases of the cell cycle.


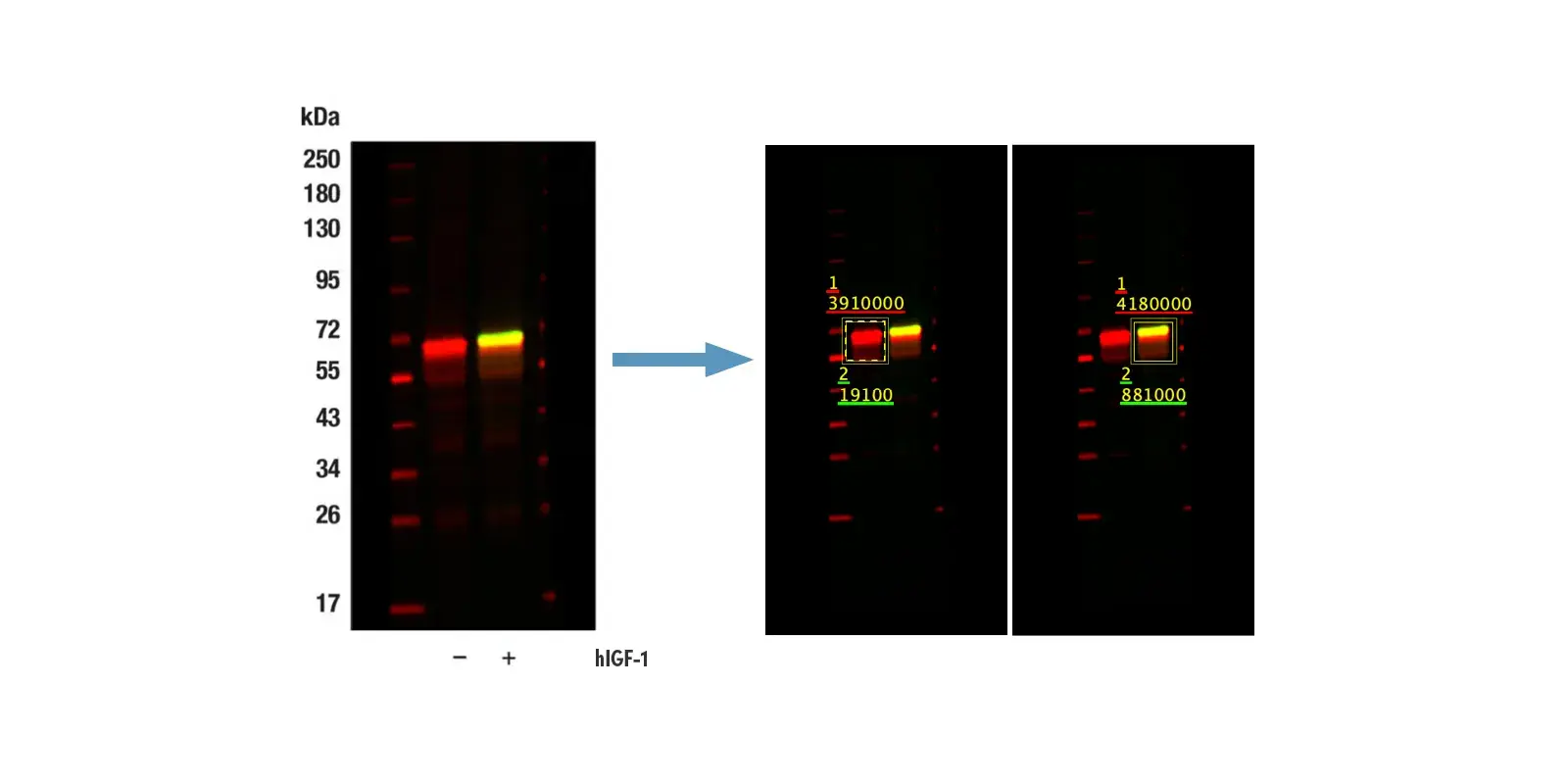
Immunohistochemical analysis of paraffin-embedded A20 syngeneic tumor using Rad51 (F4L4Y) Rabbit Monoclonal Antibody #84555.
IF analysis of HCT 116 cells (human colorectal cancer) using Rad51 (F1G6C) Rabbit mAb #65653 (green), DyLight 554 Phalloidin #13054 (red), and DAPI #4083 (blue). The presence of punctate Rad51 staining in cells is a marker for intact HR repair. - Non-Homologous End Joining (NHEJ): NHEJ is another important mechanism for repairing double-strand breaks that occurs in all phases of the cell cycle. It involves the binding of the Ku70/Ku80 complex to broken DNA ends, followed by the recruitment of monomeric DNA-PK catalytic subunits (DNA-PKcs) to form an active DNA-PK complex. Next, proteins such as Artemis, XRCC4, XLF, and DNA Ligase IV work to complete the repair process.
- Theta-Mediated End Joining (TMEJ): Generally reserved for when other repair pathways are not viable, TMEJ is an error-prone mechanism for mending double-strand breaks. DNA polymerase theta plays a crucial role in TMEJ, along with proteins including Rad51 and poly (ADP-ribose) polymerase 1 (PARP-1).



- Mismatch Repair (MMR): The mismatch repair system is composed of a complex network of proteins that recognize and repair base–base mismatches and insertion–deletion loops that arise during DNA replication. Key MMR proteins include MLH1, MSH2, MSH6, PMS2, and EXO1, many of which are mutated in human tumors. Deficiency in MMR proteins (referred to as dMMR) leads to microsatellite instability (MSI), which is associated with a predisposition to certain cancer types.


Cell Cycle Checkpoints and Regulatory Proteins
When a cell experiences DNA damage, the repair pathways discussed above are activated to fix the damage and restore genome stability. Simultaneously, the cell also engages a complex surveillance system: Cell cycle checkpoints.
Cell cycle checkpoints work at critical stages of the cell cycle—such as the G1/S and G2/M transitions—to monitor for DNA damage and coordinate the appropriate response. If DNA damage, incomplete replication, or chromosome alignment errors are detected, checkpoint proteins pause the cell cycle. This temporary arrest gives the cell time to activate DNA repair pathways before progressing to DNA replication or cell division.
If DNA damage is successfully repaired, checkpoint signaling is switched off, and the cell cycle continues. However, if the damage cannot be repaired, the checkpoints act as a second line of defense to trigger either programmed cell death (apoptosis) or permanent cell cycle arrest (senescence) to prevent potentially harmful mutations from being passed on.
Below are some of the key proteins involved in checkpoint signaling and regulation:
- ATM and ATR: Sensor kinases that activate checkpoint signaling in response to DNA double-strand breaks (ATM) and replication stress/single-stranded DNA (ATR). Their activation coordinates DNA damage detection with cell cycle arrest and repair pathway activation.
- Chk1 and Chk2: Downstream effector kinases that enforce cell cycle arrest following DNA damage. Chk1 is especially important for the S and G2/M checkpoints and stabilizing stalled replication forks, while Chk2 is activated primarily in response to double-strand DNA breaks. Both Chk1 and Chk2 can influence the homologous recombination (HR) DNA repair pathway—Chk1 can promote HR via the phosphorylation of repair proteins such as Rad51, and Chk2 can modulate HR efficiency via the phosphorylation of BRCA1.
- p53: The most commonly mutated gene in human cancers, p53 is a tumor suppressor and transcription factor activated by numerous post-translational modifications (PTMs), including phosphorylation at serine 15. A key checkpoint regulator, p53 promotes the expression of genes that drive cells into apoptosis, cell cycle arrest, or senescence in response to various types of DNA damage.
- p21: A cyclin-dependent kinase inhibitor and direct downstream effector of p53, p21 enforces cell cycle arrest following DNA damage at the G1/S and G2/M checkpoints.
- MDM2 and MDM4 (MdmX): MDM2 and MDM4 are negative regulators of p53 that are overexpressed in many human cancers. Because they inhibit p53 in distinct yet complementary ways, therapies that target both MDM2 and MDM4 are being investigated to treat cancers that express the wild-type p53 protein.3
- Mitotic Spindle Checkpoint: Also known as the spindle assembly checkpoint (SAC), the mitotic spindle checkpoint blocks cell division when chromosomes are not properly attached to spindle microtubules. Key components of spindle checkpoint signaling include: Cyclin B1, which binds CDK1 (also referred to as cdc2) to prevent entry into mitosis until the cell has checked for replication errors; Bub1, which is involved in monitoring kinetochore-microtubule interactions; and Aurora B kinase, which functions as part of the chromosomal passenger complex to monitor and correct kinetochore-microtubule misattachments and to coordinate proper chromosome segregation before cell division.






Checkpoint Responses to DNA Replication Stress
A major trigger of genomic instability in cancer is replication stress, which occurs when a cell’s DNA replication machinery stalls or breaks down—especially in rapidly dividing cells, like those in tumors. Replication stress can leave long segments of single-stranded DNA (ssDNA) exposed, which activates checkpoint signaling cascades. ssDNA regions are rapidly coated by replication protein A (RPA), which recruits and activates the ATR-ATRIP complex, followed by the engagement of checkpoint proteins like Chk1 and TopBP1.
 IF analysis of HeLa cells, untreated (left) or UV-treated (right), using Phospho-Chk1 (Ser317) (D12H3) XP® Rabbit mAb #12302 (green). Actin filaments were labeled with DY-554 phalloidin (red). Replication stress due to UV rays leads to the phosphorylation of Chk1.
IF analysis of HeLa cells, untreated (left) or UV-treated (right), using Phospho-Chk1 (Ser317) (D12H3) XP® Rabbit mAb #12302 (green). Actin filaments were labeled with DY-554 phalloidin (red). Replication stress due to UV rays leads to the phosphorylation of Chk1.
This checkpoint signaling cascade stabilizes the DNA, restarts replication, and helps to prevent dangerous double-strand breaks. For example, exposure to UV light causes DNA damage, which triggers Chk1 phosphorylation to arrest the cell cycle and promote DNA repair before cell division.
Therapeutics: PARP Inhibitors, Immune Checkpoint Inhibitors, & Small Molecules
Because tumor cells are often deficient in at least one DNA repair pathway or checkpoint, they are generally more susceptible to DNA-damaging agents than healthy cells. For this reason, platinum-based chemotherapeutic agents such as cisplatin, carboplatin, and oxaliplatin are widely used to treat cancer. By inducing DNA damage that is too extensive to repair, these drugs drive cancer cells into apoptosis.
Another successful type of therapeutic intervention involves the inhibition of PARP, an important DNA repair enzyme. PARP inhibitors are especially effective in the treatment of BRCA1/2-deficient cancers, such as some breast cancers, where they cause a synthetic lethal effect—a situation where the simultaneous disruption of two DNA repair pathways leads to cancer cell death. BRCA1 and BRCA2 are essential for homologous recombination. Cancer cells that lack functional BRCA genes become dependent on PARP to repair single-strand breaks. When PARP is inhibited in these cells, both major repair pathways are compromised, leading to the accumulation of DNA damage and, ultimately, cell death. Normal cells, which retain functional BRCA1/2 and homologous recombination, can survive PARP inhibition, making this approach highly selective for tumor cells.

IHC analysis of paraffin-embedded human ductal breast carcinoma using BRCA1 (E5S9G) Rabbit mAb #50799 performed on the Leica BOND RX. The identification of BRCA1 in this tumor means that the patient may be less likely to respond to treatment with a PARP inhibitor.
Other DNA repair enzymes being investigated for therapeutic targeting include DNA-PK and the checkpoint proteins Chk1 and Chk2. Inhibitors of WRN helicase, a member of the RecQ DNA helicase family that helps maintain genome stability through its functions in DNA replication, recombination, and repair, are also being investigated for the treatment of cancers with high microsatellite instability (MSI).
Besides these, small molecules that interfere with the p53-MDM2/MDM4 interaction are being developed, as well as those that inhibit Aurora B and CDK12. CDK12 is a transcriptional kinase that protects genomic stability by promoting the expression of long, complex DNA repair genes, including those critical for homologous recombination. Mutations or inhibition of CDK12 lead to DNA repair deficiencies and sensitize tumors to DNA-damaging therapies.
In addition, immune checkpoint inhibitors are being explored as treatments for cancers with high mutation burdens, such as those associated with mismatch repair deficiency (dMMR) and MSI, conditions caused by defects in the MMR system that make cancer cells more susceptible to mutation.
Future Directions for Cancer Therapy Targeting Genomic Instability
Given the complexity of cancer, researchers must continue to develop innovative therapies. Strategies being pursued include synthetic lethal approaches beyond PARP inhibitors, and the combination of DNA repair inhibitors with other therapies, including inhibitors of cell cycle progression. It is also increasingly common to use liquid biopsy to examine circulating tumor DNA (ctDNA) for mutations in key DNA repair proteins, which would inform patient stratification and enable personalized medicine.
The more we understand about cancer and its underlying mechanisms, the closer we get to a cure.
Additional Resources
Read the additional blogs in the Hallmarks of Cancer Series:
- Evading Growth Suppressors
- Nonmutational Epigenetic Reprogramming
- Avoiding Immune Destruction
- Tumor-Promoting Inflammation
- Activating Invasion & Metastasis
- Inducing or Accessing Vasculature (Angiogenesis)
- Senescent Cells
- Resisting Cell Death
- Deregulating Cellular Metabolism
- Unlocking Phenotypic Plasticity
- Sustaining Proliferative Signaling
Select References
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57-70. doi:10.1016/s0092-8674(00)81683-9
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-674. doi:10.1016/j.cell.2011.02.013
- Toledo F, Wahl GM. MDM2 and MDM4: p53 regulators as targets in anticancer therapy. Int J Biochem Cell Biol. 2007;39(7-8):1476-1482. doi:10.1016/j.biocel.2007.03.022
- Yates LA, Zhang X, Burgers PM. DNA Damage and Replication Stress Checkpoints. Annu Rev Biochem. 2025;94(1):195-221. doi:10.1146/annurev-biochem-072324-031915
- Jordan MR, Mendoza-Munoz PL, Pawelczak KS, Turchi JJ. Targeting DNA damage sensors for cancer therapy. DNA Repair (Amst). 2025;149:103841. doi:10.1016/j.dnarep.2025.103841
- Qian J, Liao G, Chen M, et al. Advancing cancer therapy: new frontiers in targeting DNA damage response. Front Pharmacol. 2024;15:1474337. Published 2024 Sep 20. doi:10.3389/fphar.2024.1474337
- Vaddavalli PL, Schumacher B. The p53 network: cellular and systemic DNA damage responses in cancer and aging. Trends Genet. 2022;38(6):598-612. doi:10.1016/j.tig.2022.02.010
- Petsalaki E, Zachos G. DNA damage response proteins regulating mitotic cell division: double agents preserving genome stability. FEBS J. 2020;287(9):1700-1721. doi:10.1111/febs.15240
25-HMC-24850