Inflammation can play different roles in the context of cancer. Short-lived inflammatory signaling can promote the clearance of damaged cells, including cancerous and pre-cancerous cells. When inflammatory signals fail to appropriately resolve, however, the resulting chronic inflammatory environment can directly and indirectly promote cancer formation, survival, and metastasis.
< Jump to the product list at the end of this blog >
|
This post explores many of the cell types and signaling molecules involved in the cancer hallmark Tumor-Promoting Inflammation. It also highlights key pathways and cell markers that can be used to study this enabling characteristic of cancer. Relevant Pathway DiagramsCancer cells alter cellular signaling to promote inflammatory cues in the tumor microenvironment (TME) and drive genomic instability, inhibit apoptosis, limit anti-tumor immune responses, and stimulate angiogenesis. |
 Tumor-Promoting Inflammation was first described in 2011 as an enabling characteristic—a risk factor that can promote the six original hallmarks of cancer identified in 2000.
Tumor-Promoting Inflammation was first described in 2011 as an enabling characteristic—a risk factor that can promote the six original hallmarks of cancer identified in 2000.The following pathways are most relevant to Tumor-Promoting Inflammation:
 |
Inflammasome Signaling PathwayInflammasomes, such as the NLRP3 inflammasome, form in response to infection- or damage-associated signals and activate caspase‑1 to process IL‑1β and IL‑18. Persistent activation can sustain inflammation and tissue damage that supports tumor development. Get the Inflammasome Signaling Pathway diagram |
 |
Jak/Stat: IL-6 Receptor Signaling PathwayIL‑6 receptor engagement activates Jak kinases and Stat transcription factors, including Stat3, to drive survival, proliferation, and inflammatory gene expression. Chronic IL‑6/Jak/Stat3 signaling links inflammation to tumor growth and immune evasion. Get the Jak/Stat IL-6 Receptor Signaling Pathway diagram |
 |
NF‑κB Signaling PathwayIn the NF‑κB pathway, phosphorylation and degradation of IκBα frees NF‑κB to enter the nucleus and induce expression of cytokines, chemokines, and pro‑survival genes. Sustained NF‑κB activation in tumors promotes proliferation, blocks apoptosis, and maintains an inflammatory TME. |
Acute vs Chronic Inflammation: What’s the Difference?
Acute inflammation generally represents a healthy physiological response to injury or infection, resulting in pathogen clearance, wound healing, and, in some cases, anti-tumor immune response. In contrast, chronic inflammation is generally deleterious and can promote tumorigenesis by sustaining an environment rich in cytokines, growth factors, and DNA-damaging species. The best way to differentiate between acute and chronic inflammation is through observation over time—while acute inflammation resolves quickly, chronic inflammation can last for weeks, months, or even indefinitely.
During acute inflammation, neutrophils are rapidly recruited to sites of tissue damage or infection. In chronic inflammation, additional myeloid subsets, such as myeloid‑derived suppressor cells (MDSCs), expand and persist, contributing to sustained inflammatory signaling and tissue remodeling.

Key Cell Types in the Inflammatory Response
During the general course of inflammation, neutrophils (Human markers: CD11b/ITGAM (pan-myeloid), CD16, CD15/SSEA, CD66b, CD10 (mature); Mouse: CD11b/ITGAM (pan-myeloid), Ly-6G) serve as first responders to inflammatory cues such as the pathogen- and danger-associated molecular patterns (PAMPs/DAMPs) released in response to tissue injury and infection. Neutrophils clear pathogens and necrotic cells at inflamed sites via phagocytosis and the release of antimicrobial mediators. They also secrete factors that directly and indirectly (via endothelial activation) recruit circulating monocytes to the tissue.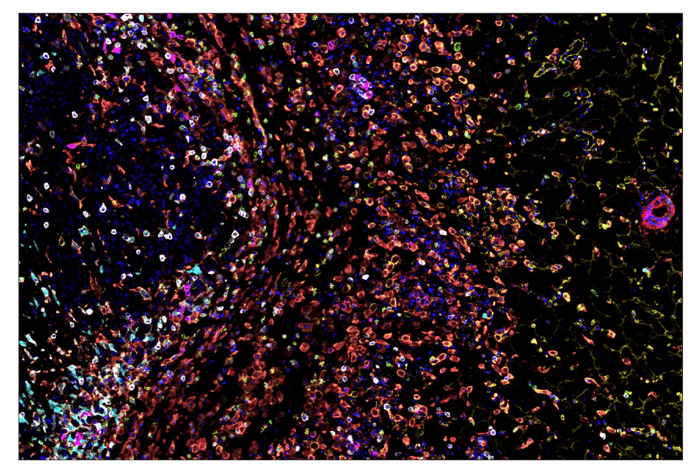
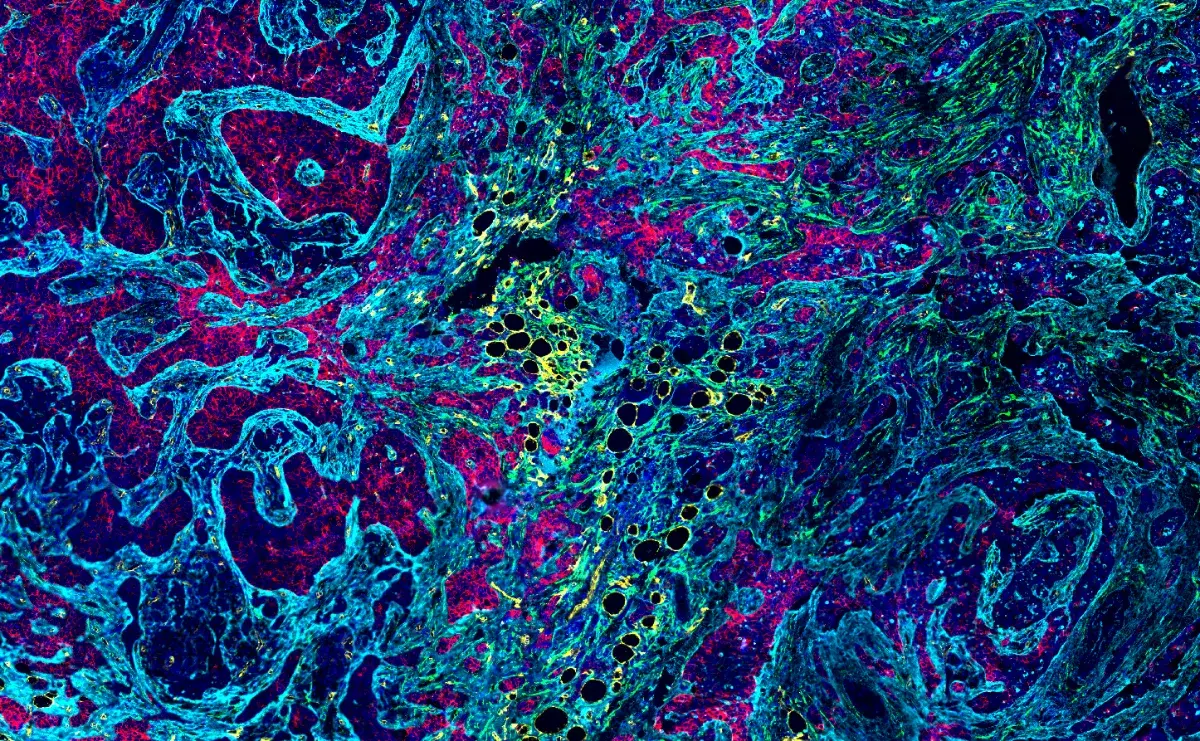
SignalStar® multiplex immunohistochemical analysis of paraffin-embedded GL261 syngeneic tumor using CD11b/ITGAM (E4K8C) & CO-0083-488 SignalStar Oligo-Antibody Pair #39172 (green), CD36 (E8B7S) & CO-0089-594 SignalStar Oligo-Antibody Pair #23418 (yellow), Pan-Keratin (Type I) (E6S1S) & CO-0072-647 SignalStar Oligo-Antibody Pair #77030 (red), IL-2Rα/CD25 (E9W2J) & CO-0074-750 SignalStar Oligo-Antibody Pair #71915 (magenta), CD8 alpha (D4W2Z) & CO-0040-488 SignalStar Oligo-Antibody Pair #17707 (white), Arginase-1 (D4E3M) & CO-0075-594 SignalStar Oligo-Antibody Pair #66757 (cyan), F4/80 (D2S9R) & CO-0042-750 SignalStar Oligo-Antibody Pair #51924 (orange), and DAPI #4083 (blue). All fluorophores have been assigned a pseudocolor, as indicated. Staining was performed on the BOND RX autostainer by Leica Biosystems.
Recruited monocytes differentiate into macrophages (Human markers: CD68, HLA‑DR; Mouse: F4/80), which can adopt a number of activation states in response to different signaling cues, generally categorized as pro-inflammatory, M1-like (pro-inflammatory; human/mouse markers: CD80, CD86) or M2‑like (pro‑resolving/tissue‑repair; human/mouse markers: CD206/MRC1, CD163) phenotypes. M1-like macrophages help clear pathogens and tumor cells and can induce neutrophil apoptosis, whereas M2‑like macrophages exhibit enhanced efferocytic capacity, clear dead neutrophils, and support tissue repair and remodeling. If the balance between these two functional states is disrupted, the inflammatory response can fail to resolve, leading to chronic inflammation characterized by persistent M1‑ and M2‑like macrophages and ongoing neutrophil recruitment.
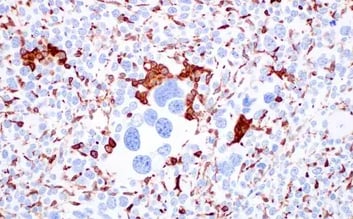
How Inflammation Contributes to Cancer Initiation and Progression
Promoting Genomic Instability & Damaging DNA Repair Mechanisms
One of the main ways that inflammation contributes to cancer initiation is by promoting genomic instability and mutation, another cancer hallmark, often through the production of reactive oxygen species (ROS) at sites of chronic inflammation. Under normal conditions, both neutrophils and macrophages produce large quantities of ROS as part of the antimicrobial response, and these molecules help drive M1-like macrophage differentiation, inflammatory cytokine release (for example, IL‑1β, TNF‑α, and IFN-β), and, at high concentrations, apoptosis.
In chronically inflamed tissues, however, persistent ROS exposure induces DNA strand breaks, base modifications, and other lesions that can overwhelm DNA repair mechanisms and lead to mutagenesis. Chronic exposure to inflammatory mediators can also directly disrupt DNA repair pathways—altering the expression or activity of key repair proteins—which further compromises genome maintenance and allows damaged cells to persist. Over time, this accumulation of DNA damage increases the risk that cells will acquire oncogenic mutations and undergo malignant transformation.
Promoting Cell Proliferation
Inflammation also promotes cell proliferation, which can further expand cells carrying oncogenic mutations. At moderate concentrations, ROS fosters NF‑κB activation by inducing phosphorylation and degradation of IκBα, which typically sequesters NF-κB outside the nucleus in an inactive state. When IκBα is degraded, NF-κB is able to enter the nucleus to drive the expression of genes that support proliferation, survival, and the production of inflammatory cytokines.
 |
 |
|
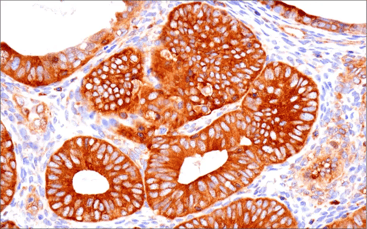
IHC analysis of paraffin-embedded human endometrioid carcinoma using NF-κB p65 (D14E12) Rabbit Monoclonal Antibody #8242. |
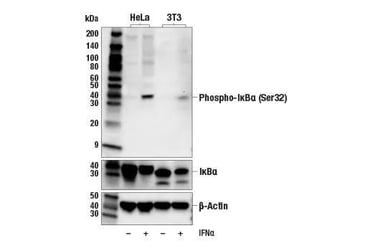
Western blot analysis of extracts from various cell lines, untreated or treated with IFN-α using Phospho-IκBα (Ser32) (14D4) Rabbit mAb #2859 (upper), IκBα (L35A5) Mouse mAb (Amino-terminal Antigen) #4814 (middle), and β-Actin (D6A8) Rabbit mAb #8457 (lower). |
Several pro-inflammatory cytokines and chemokines released at sites of inflammation also directly promote cell proliferation and survival. IL‑6 binding to the IL‑6 receptor activates Jak2 and Stat3, leading to transcription of Stat3-regulated genes that drive cell-cycle progression and anti-apoptotic programs. IL‑1β and TNF‑α signal through their respective receptors to trigger IκBα phosphorylation and degradation and derepress NF‑κB, further reinforcing proliferation and inflammatory signaling.
 |
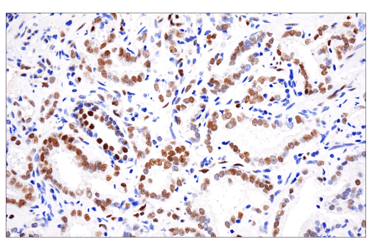 |
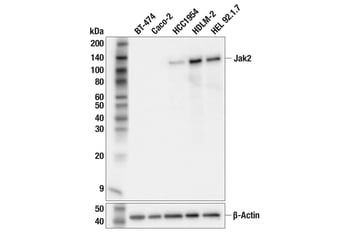
| WB extracts from various human cell lines using Jak2 (F7S5R) Rabbit Monoclonal Antibody #65483 (upper) or beta-Actin (D6A8) Rabbit Monoclonal Antibody #8457 (lower). Negative expression of Jak2 protein in BT-474 and Caco-2 cells is consistent with the predicted expression pattern. | IHC analysis of paraffin-embedded human prostate adenocarcinoma using Phospho-Stat3 (Tyr705) (D3A7) Rabbit Monoclonal Antibody #9145 performed on the BOND RX autostainer by Leica Biosystems. |
Persistent Microbial Infection, Inflammation, and Cancer
An additional route by which inflammation initiates cancer involves persistent microbial infection. Chronic infection can sustain activation of pattern recognition receptors and inflammasomes, including the NLRP3 inflammasome, which assembles around the adaptor ASC/TMS1 to recruit and activate caspase-1, driving production and activation of IL‑1β and other proinflammatory cytokines. Colitis‑associated colorectal cancer and Helicobacter pylori–induced gastric cancer are classic examples where increased bacterial burden, virulence factors, and ongoing tissue injury are linked to DNA damage and prolonged activation of NF‑κB, Stat3, and the Wnt signaling pathway.
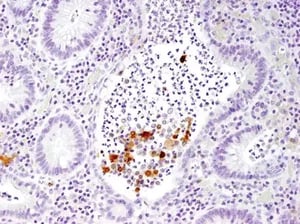 |
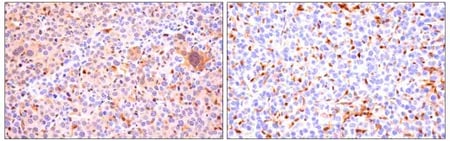 |
|
IHC analysis of paraffin-embedded human large intestine (chronic colitis of the colon) using IL-1 beta (3A6) Mouse Monoclonal Antibody #12242. |
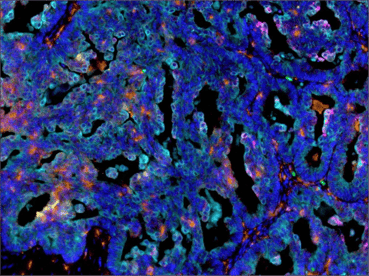
IHC analysis of paraffin-embedded Renca syngeneic tumor (top), 4T1 syngeneic mammary tumor (right) using ASC/TMS1 (D2W8U) Rabbit Monoclonal Antibody #67824. Both tumors show staining of infiltrating immune cells. Note the presence of staining in the Renca tumor cells and the lack of staining in the 4T1 tumor cells. |
Patients with inflammatory bowel disease (IBD), especially ulcerative colitis, are up to twice as likely to develop colon cancer as the general population due to the cumulative impact of chronic mucosal inflammation and genomic damage. Similarly, studies indicate that the majority of gastric cancers can be attributed to chronic H. pylori infection, underscoring the carcinogenic potential of long-term, infection-associated inflammation.
Tumor Growth, Survival, & Metastasis: How Inflammation Fuels Existing Cancer
Once malignant cells are established, inflammation can drive disease progression by promoting tumor growth, survival, immune escape, and spread. Within the TME, several myeloid populations—including M2 tumor‑associated macrophages (M2 TAMs), N2 tumor‑associated neutrophils (N2 TANs), and MDSCs, rapidly expand during emergency myelopoiesis to create a pro-tumor, immunosuppressive niche. These cells dampen anti‑tumor T cell activity by depleting arginine via arginase‑1 enzymatic activity, secreting immunosuppressive cytokines such as IL‑10 and TGF‑β, and expressing immune checkpoint proteins such as PD‑L1 and CTLA‑4. They also release VEGF‑A and MMP‑9, which drive extracellular matrix remodeling, angiogenesis, and local invasion.
 IHC analysis of paraffin-embedded human colon adenocarcinoma using Arginase-1 (D4E3M) Rabbit Monoclonal Antibody #93668 performed on the BOND RX autostainer by Leica Biosystems.
IHC analysis of paraffin-embedded human colon adenocarcinoma using Arginase-1 (D4E3M) Rabbit Monoclonal Antibody #93668 performed on the BOND RX autostainer by Leica Biosystems.
Inflammation also enables cancer progression by inducing neutrophil extracellular traps (NETs) in and around the TME. NETs, commonly identified by citrullinated histone H3, are web-like structures composed of DNA, histones, and granule proteins. They normally trap and kill microbes, but in tumors, they can enhance metastasis by promoting epithelial–mesenchymal transition (EMT), capturing circulating tumor cells, and facilitating breakdown of the extracellular matrix (ECM).
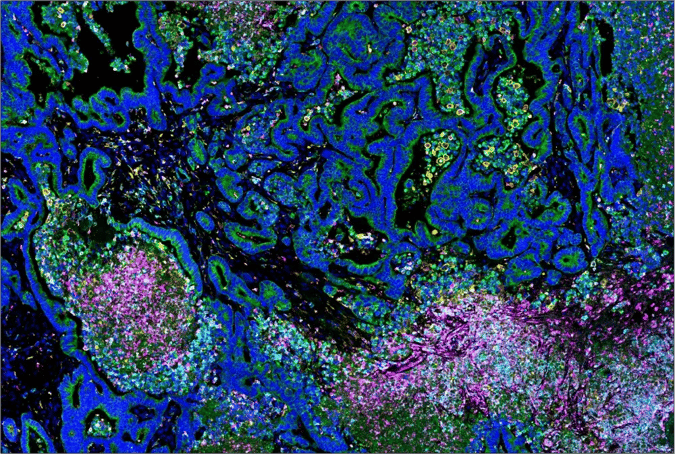
SignalStar® oligo-based multiplex IHC analysis of paraffin-embedded human colon carcinoma using Citrullinated Histone H3 (Arg17) (E4O3F) & CO-0201-488 SignalStar Oligo-Antibody Pair #43376 (488; magenta), SIRPα/SHPS1 (D6I3M) Rabbit Monoclonal Antibody #60004 (594; cyan), S100A9 (D5O6O) Rabbit Monoclonal Antibody #43317 (647; red), CD163 (D6U1J) Rabbit Monoclonal Antibody #43547 (594; yellow), Pan-Keratin (C11) Mouse Monoclonal Antibody #16373 (647; green), and DAPI #4083 (blue). Staining was performed on the BOND RX autostainer by Leica Biosystems.
Another layer of complexity comes from chronic activation of the cGAS–STING pathway in cancers with chromosomal instability. Cytosolic DNA derived from micronuclei or damaged chromosomes can activate cGAS–STING signaling, leading to production of inflammatory cytokines and type I interferons. While transient cGAS-STING activation enhances anti-tumor immunity, sustained activation, which often occurs in chronic inflammatory settings, can help establish an immunosuppressive, tumor-promoting microenvironment.
Tipping the Scale: Bringing Tumor-Promoting Inflammation Back into Balance
The role of inflammation in cancer is complex and highly context-dependent, with acute responses sometimes supporting immune surveillance and tumor elimination, and chronic responses often driving tumor initiation and progression.
A number of interventions aimed at combating chronic inflammation are under investigation for use as cancer treatments. These include monoclonal antibody and small-molecule drugs targeting IL‑1β, IL‑6, their receptors, and the Jak/Stat pathway. Many of these drugs, such as tocilizumab and ruxolitinib, are already clinically approved to treat inflammatory conditions like rheumatoid arthritis and atopic dermatitis.
Finding additional ways to selectively reprogram or modulate inflammatory pathways and myeloid cell states in the TME will be critical for future therapeutic strategies that aim to prevent cancer development and improve responses to existing treatments. Efforts to selectively target tumor MDSCs for apoptosis and to promote conversion of pro-tumor M2-like macrophages into anti-tumor M1-like macrophages are ongoing.
Current research emphasizes the importance of restoring a healthy balance—resolving chronic, tumor-promoting inflammation while preserving the body’s ability to mount robust immune responses to infection and emerging tumor cells. Understanding how to modulate patients’ inflammatory responses to strike an appropriate balance will likely remain a key focus area for the foreseeable future.
| Product | Applications | Reactivity |
| CD11b/ITGAM (D6X1N) & CO-0037-647 SignalStar Oligo-Antibody Pair #29052 | SignalStar IHC | H |
| CD11b/ITGAM (E4K8C) & CO-0083-594 SignalStar Oligo-Antibody Pair #84898 | SignalStar IHC | M |
| Citrullinated Histone H3 (Arg17) (E4O3F) & CO-0201-488 SignalStar Oligo-Antibody Pair #43376 | SignalStar IHC | H, M |
| STING (D2P2F) & CO-0219-647 SignalStar Oligo-Antibody Pair #33694 | SignalStar IHC | H |
| Human Reactive Inflammatory Cytokine Antibody Sampler Kit #83636 | ||
| NF-kappaB Pathway Antibody Sampler Kit #9936 | ||
| Stat3/Stat5 Signaling Antibody Sampler Kit #67088 |
Additional Resources
Read the additional blogs in the Hallmarks of Cancer Series:
- Evading Growth Suppressors
- Nonmutational Epigenetic Reprogramming
- Avoiding Immune Destruction
- Activating Invasion & Metastasis
- Inducing or Accessing Vasculature (Angiogenesis)
- Senescent Cells
- Genome Instability & Mutation
- Resisting Cell Death
- Deregulating Cellular Metabolism
- Unlocking Phenotypic Plasticity
- Sustaining Proliferative Signaling
Select References
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57-70. doi:10.1016/s0092-8674(00)81683-9
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-674. doi:10.1016/j.cell.2011.02.013
- Hanahan D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022;12(1):31-46. doi:10.1158/2159-8290.CD-21-1059
- Kay J, Thadhani E, Samson L, Engelward B. Inflammation-induced DNA damage, mutations and cancer. DNA Repair (Amst). 2019;83:102673. doi:10.1016/j.dnarep.2019.102673
- Wu L, Zhang XH. Tumor-Associated Neutrophils and Macrophages-Heterogenous but Not Chaotic. Front Immunol. 2020;11:553967. Published 2020 Dec 2. doi:10.3389/fimmu.2020.553967
- Li K, Shi H, Zhang B, et al. Myeloid-derived suppressor cells as immunosuppressive regulators and therapeutic targets in cancer. Signal Transduct Target Ther. 2021;6(1):362. Published 2021 Oct 7. doi:10.1038/s41392-021-00670-9
- Nishida A, Andoh A. The Role of Inflammation in Cancer: Mechanisms of Tumor Initiation, Progression, and Metastasis. Cells. 2025;14(7):488. Published 2025 Mar 25. doi:10.3390/cells14070488
- Su Y, Gao J, Kaur P, Wang Z. Neutrophils and Macrophages as Targets for Development of Nanotherapeutics in Inflammatory Diseases. Pharmaceutics. 2020;12(12):1222. Published 2020 Dec 17. doi:10.3390/pharmaceutics12121222
- Greten FR, Grivennikov SI. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity. 2019;51(1):27-41. doi:10.1016/j.immuni.2019.06.025
- Wang Y, Yi P, Chen Y, et al. Inflammation and tumor progression: signaling pathways and targeted intervention. Signal Transduct Target Ther. 2021;6(1):387. doi:10.1038/s41392-021-00658-5.
- Aggarwal V, Tuli HS, Varol A, et al. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules. 2019;9(11):735. Published 2019 Nov 13. doi:10.3390/biom9110735
- Hong Y, Boiti A, Vallone D, Foulkes NS. Reactive Oxygen Species Signaling and Oxidative Stress: Transcriptional Regulation and Evolution. Antioxidants (Basel). 2024;13(3):312. Published 2024 Mar 1. doi:10.3390/antiox13030312
- Lin TY, Tsai MC, Tu W, et al. Role of the NLRP3 Inflammasome: Insights Into Cancer Hallmarks. Front Immunol. 2021;11:610492. Published 2021 Feb 3. doi:10.3389/fimmu.2020.610492
- Shadab A, Mahjoor M, Abbasi-Kolli M, Afkhami H, Moeinian P, Safdarian AR. Divergent functions of NLRP3 inflammasomes in cancer: a review. Cell Commun Signal. 2023;21(1):232. Published 2023 Sep 15. doi:10.1186/s12964-023-01235-9
- Tengesdal IW, Dinarello CA, Marchetti C. NLRP3 and cancer: Pathogenesis and therapeutic opportunities. Pharmacol Ther. 2023;251:108545. doi:10.1016/j.pharmthera.2023.108545
- Dan WY, Zhou GZ, Peng LH, Pan F. Update and latest advances in mechanisms and management of colitis-associated colorectal cancer. World J Gastrointest Oncol. 2023;15(8):1317-1331. doi:10.4251/wjgo.v15.i8.1317
- Salvatori S, Marafini I, Laudisi F, Monteleone G, Stolfi C. Helicobacter pylori and Gastric Cancer: Pathogenetic Mechanisms. Int J Mol Sci. 2023;24(3):2895. Published 2023 Feb 2. doi:10.3390/ijms24032895
- Sanchez-Pino MD, Dean MJ, Ochoa AC. Myeloid-derived suppressor cells (MDSC): When good intentions go awry. Cell Immunol. 2021;362:104302. doi:10.1016/j.cellimm.2021.104302
- Wang Y, Yang K, Li J, Wang C, Li P, Du L. Neutrophil extracellular traps in cancer: From mechanisms to treatments. Clin Transl Med. 2025;15(6):e70368. doi:10.1002/ctm2.70368
- Yue B, Gao W, Lovell JF, Jin H, Huang J. The cGAS-STING pathway in cancer immunity: dual roles, therapeutic strategies, and clinical challenges. Essays Biochem. 2025;69(2):EBC20253006. Published 2025 Mar 7. doi:10.1042/EBC20253006