Cancer thrives by keeping cell proliferation stuck in the “on” position. Normally, cells wait for the green light from cytokines, growth factors, and regulatory checkpoints to proceed through division. Cancer cells, however, bypass these checks and balances by rewiring growth pathways, allowing them to ignore stop signals and grow uncontrollably.
One of the original Hallmarks of Cancer, Sustaining Proliferative Signaling captures the idea that tumor cells become increasingly independent of normal external growth controls. In this post, we take a closer look at how major growth pathways are altered in cancer, and highlight specific receptors, kinases, and cell cycle regulators being targeted for therapeutic intervention.
< Jump to the product list at the end of this blog >
Key Signaling Pathways: How Cancer Hacks the “On” Switch
The RAS-RAF-MEK-ERK and PI3K/AKT signaling pathways are critical for controlling cell growth and are the most commonly hijacked growth signaling networks in cancer. Their dysregulation can result from mutations in key pathway components or from abnormal activation of upstream cell surface receptors, such as receptor tyrosine kinases (RTKs) like ALK and ROS1.
|
Both signaling pathways are shaped by complex feedback loops and cross-talk between pathway components, which can further amplify or dampen growth signals. Because the RAS-RAF-MEK-ERK and PI3K/AKT pathways frequently intersect and share common nodes, such as PI3 kinase (PI3K), cancer cells can often reroute signaling through alternative receptors or branches of the network, allowing proliferation to continue even when a drug blocks one receptor or arm of the pathway. |
What are the Hallmarks of Cancer?The Hallmarks of Cancer1-3 are a research framework that organizes the key traits cancer cells acquire in order to grow and spread. Initially described by Douglas Hanahan and Robert Weinberg in 2000, the framework groups the underlying mechanisms of cancer into a series of smaller subsets to advance discovery. The concept was expanded in 2011 to include two additional hallmarks and two enabling characteristics, and again in 2022 with four new emerging hallmarks.
|
 Sustaining Proliferative Signaling is one of the original cancer hallmarks, first described in 2000.
Sustaining Proliferative Signaling is one of the original cancer hallmarks, first described in 2000.This redundancy makes cancer signaling highly complex and is a major reason why developing effective, targeted therapies remains challenging.
RAS-RAF-MEK-ERK Pathway
|
|
Download the MAPK/Erk in Growth and Differentiation pathway diagram, including key CST antibody products. |
 |
Download the G-Protein-Coupled Receptors Signaling to MAPK/Erk pathway diagram, including key CST antibody products. |

PI3K/AKT and mTOR Pathways
|
|
Download the PI3K/AKT Signaling pathway diagram, including key CST antibody products. |
Receptor Tyrosine Kinases: Where Turning “On” Abnormal Growth Starts
Receptor tyrosine kinases (RTKs) are cell-surface receptor proteins that act as molecular antennas, sensing growth signals and relaying them inward by phosphorylating tyrosines on themselves and other proteins. This mechanism ensures cells respond appropriately to external growth cues.
In cancer cells, however, RTKs often become chronically active and bypass normal regulation. RTKs that drive cell proliferation include the ALK, EGFR, HER2, ROS1, and FGF receptors.
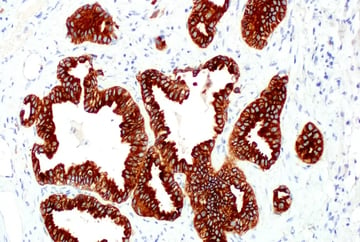
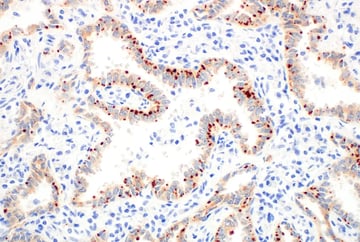
After binding a ligand, RTKs undergo phosphorylation at their cytoplasmic tails (C-termini), which initiates the intracellular signaling. Oncogenic mutations and gene fusions involving RTK kinase domains—for example, CD74‑ROS1, SLC34A2‑ROS1, EML4‑ALK, and NPM‑ALK—can create abnormally active receptors that drive tumor growth. Additionally, in some types of cancer, RTK domains can become uncoupled from the membrane receptor and instead signal in the cytoplasm, allowing them to remain active without growth factor binding or Ras activation.
RAS-RAF-MEK-ERK Signaling and Cancer
The RAS-RAF-MEK-ERK pathway plays a central role in determining when cells should grow and divide by conveying growth signals from activated RTKs on the cell surface to the nucleus. This starts a signaling cascade inside the cell involving RAS, RAF, MEK1/MEK2, and ERK1/2 (also called p44/p42 MAPK). Once activated via phosphorylation at Thr202/Tyr204, ERK enters the nucleus, where it turns on transcription factors such as c-Myc, c-Fos, and members of the ETS family, which transcribe genes that drive cell division.
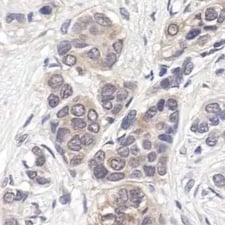
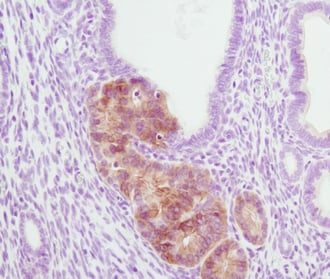
IHC analysis of paraffin-embedded human breast carcinoma, showing cytoplasmic and nuclear localization, using p44/42 MAPK (Erk1/2) (137F5) Rabbit Monoclonal Antibody #4695.
In cancer, however, persistent activation in key proteins such as RAS or RAF can mean that the “grow” signal never gets shut off, leading to uncontrolled cell division.
RAS activation is tightly controlled by Ras-Guanine Nucleotide Exchange Factors (GEFs), such as SOS1, and Ras-GTPase Activating Proteins (GAPs), including RASA2 and RASAL1/RASAL2. Mutations in RAS, including Ras G12V, Ras G12D, and Ras G12C, lock the protein in its active form by preventing GDP-bound deactivation, driving continuous downstream signaling.
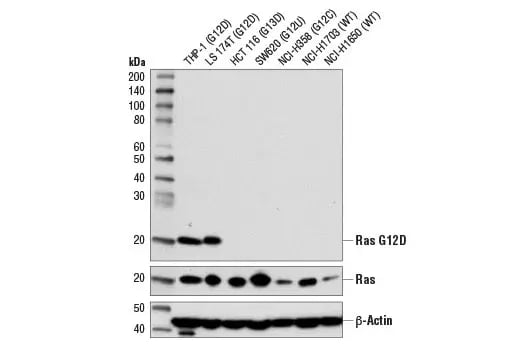
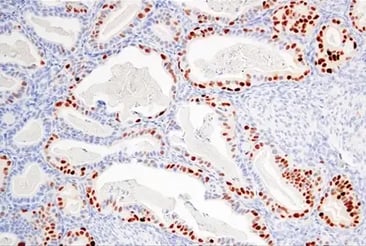
Western blot analysis of extracts from various cell lines using Ras (G12D Mutant Specific) (D8H7) Rabbit Monoclonal Antibody #14429 (upper), Ras (D2C1) Rabbit Monoclonal Antibody #8955 (middle), and beta-Actin (D6A8) Rabbit Monoclonal Antibody #8457 (lower).
Active RAS recruits and activates downstream RAF protein kinases—A-Raf, B-Raf, and C-Raf—each of which contains multiple regulatory phosphorylation sites. When active, Raf proteins form dimers and heterodimers that activate MEKs. RAF activity is further modulated by phosphorylation by Akt and via mutations, such as BRAF V600E, which can increase kinase activity. These changes can lead to unchecked MEK-ERK activation, and potentially trigger a cascade of signaling events and sustained activation of growth-promoting transcriptional programs.
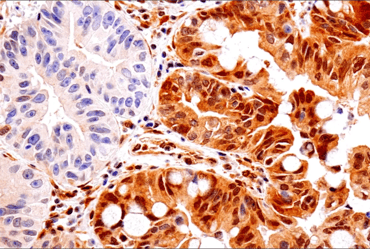
 IHC analysis of paraffin-embedded human colon carcinoma (left, B-Raf V600E positive) and human colon carcinoma (right, B-Raf V600E negative) using B-Raf (V600E Mutant) (IHC600) Mouse mAb #29002. Mutational status confirmed by genomic sequencing.
IHC analysis of paraffin-embedded human colon carcinoma (left, B-Raf V600E positive) and human colon carcinoma (right, B-Raf V600E negative) using B-Raf (V600E Mutant) (IHC600) Mouse mAb #29002. Mutational status confirmed by genomic sequencing.
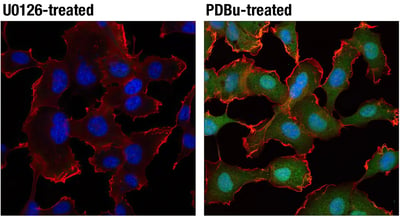
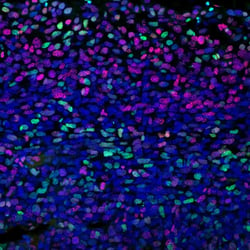
Experimental Design Tip: How to Activate MEK & ERK SignalingThe RAS-RAF-MEK-ERK pathway can be experimentally manipulated by stimulating MEK1/2–ERK1/2 signaling with RTK growth factors or Protein Kinase C (PKC) agonists such as TPA or PDBu, or by blocking it with MEK1/2 inhibitors like U0126. IF analysis of HT1080 cells, starved overnight then treated with U0126 #9903 (10 µM, 2 h; left) or PDBu (Phorbol 12,13-Dibutyrate) #12808 (100 nM, 15 m; right) using Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (D13.14.4E) Rabbit Monoclonal Antibody #4370 (green) and β-Actin (8H10D10) Mouse mAb #3700 (red). Blue pseudocolor = DRAQ5® #4084 (fluorescent DNA dye). |
PI3K/Akt and mTOR Pathways and Cancer
The PI3K/AKT pathway is initiated when growth factors outside the cell or other upstream signals activate PI3K at the cell surface. PI3K then converts the membrane lipid PIP2 into PIP3, and PIP3 subsequently binds to the pleckstrin homology (PH) domains of Akt and its upstream kinase, PDK1, recruiting both to the cell membrane for activation. At the membrane, PDK1 phosphorylates Akt at Thr308, and the mTORC2 complex phosphorylates Akt at Ser473, which together fully activate Akt. Once fully active, Akt then engages key downstream effectors, most notably the mTOR complexes, to promote cell survival, growth, and metabolism while suppressing pro‑apoptotic signals.
mTOR functions through two distinct complexes, mTORC1 and mTORC2, which connect growth signals to cell metabolism and proliferation. mTORC2, composed of mTOR, Rictor, Sin1, and Protor-1/Protor-2, acts at this early stage of the pathway to complete Akt activation in a PI3K/PIP3‑dependent manner, as described above. This makes mTORC2 part of a feed‑forward loop in which PI3K‑generated PIP3 not only recruits Akt and PDK1, but also promotes mTORC2 activity that reinforces PI3K/Akt signaling. mTORC1, which contains mTOR, GβL, and the defining subunit Raptor, drives protein synthesis and cell growth via effectors such as S6 kinase. mTORC1 activity is negatively regulated by the Hamartin/TSC1-Tuberin/TSC2 complex, and by inhibitory binding partners including DEPTOR/DEPDC6, PRAS40, and FKBP8. The subunits DEPTOR/DEPDC6 and GβL associate with both mTORC1 and mTORC2 assembly and activity, helping connect growth factor inputs to changes in Akt signaling.
PI3K and the tumor suppressor PTEN act as opposing regulators of Akt signaling. By dephosphorylating PIP3 back to PIP2, PTEN reverses the lipid signal generated by PI3K, thereby serving as a brake on this pathway by limiting membrane recruitment and activation of Akt and its downstream effectors, including mTORC1 and mTORC2. In many cancers, mutations in PI3K, AKT, or loss of PTEN function cause the pathway to be continually switched on. The persistent production of PIP3 maintains active Akt, which drives uncontrolled proliferation.
Akt also phosphorylates tumor suppressors such as p21 (Waf1/Cip1) and p27Kip1, disabling their ability to inhibit cyclin–CDK complexes—the main “engines” that drive progression through the cell cycle. For example, Akt phosphorylation of p21 at Thr145 weakens its interaction with CDKs and PCNA, reducing its ability to slow cell‑cycle progression. Similarly, Akt phosphorylation of p27Kip1 at Thr198 promotes its binding to 14‑3‑3 proteins and traps p27 in the cytoplasm, keeping it away from cyclin–CDK complexes in the nucleus.
Finally, activated Akt also inactivates glycogen synthase kinase‑3 (GSK‑3), a kinase that normally inhibits cyclin D1 and other pro‑growth signals.
Together, these changes remove important brakes on the cell cycle, allowing cells to divide more rapidly and escape normal growth controls.
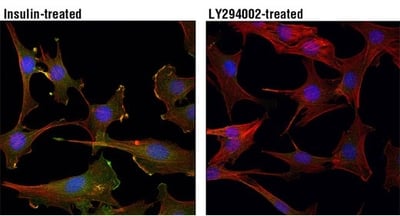
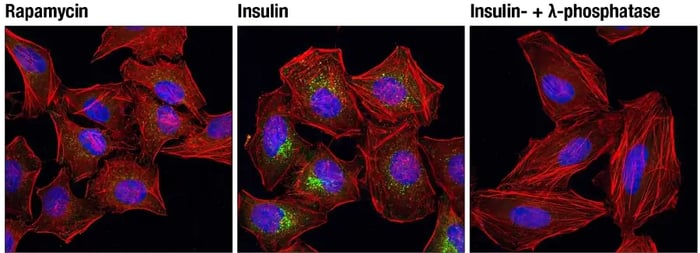
Experimental Design Tips: Activating Akt PhosphorylationThe PI3K/Akt pathway can be experimentally manipulated by adding growth factors (such as insulin, IGF-1, or EGF) to activate Akt or by using PI3K inhibitors, such as LY294002 or wortmannin, to block the formation of PIP3 and prevent Akt activation. IF analysis of C2C12 cells, insulin-treated (100 nM, 15 min; left) or treated with LY294002 #9901 (50 μM, 2 hr; right), using Phospho-Akt (Thr308) (D25E6) Rabbit Monoclonal Antibody #13038 (green). Red = Actin. Blue = DNA. Manipulating mTORTo activate mTORC1, stimulate upstream pathways that signal through PI3K/Akt, for example, by adding growth factors or nutrients (such as insulin, IGF‑1, or amino acids), which promote mTORC1‑dependent control of cell growth and metabolism. mTORC1 is acutely sensitive to rapamycin and can be inhibited using rapamycin or rapalog compounds to reduce protein synthesis and cell growth. In contrast, mTORC2 is less sensitive to short‑term rapamycin treatment. IF analysis of HeLa cells, rapamycin-treated (#9904, 10 nM for 2 hours, left), insulin-treated (150 nM for 6 minutes, middle) or insulin- and λ-phosphatase-treated (right), using Phospho-mTOR (Ser2448) (D9C2) Rabbit Monoclonal Antibody #5536 (green). Red = Actin. Blue = DNA. |
Therapeutic Interventions: Shutting Cancer Off by Targeting “On” Signaling
Multiple targeted therapies have been developed to block aberrant proliferative signaling by inhibiting receptor tyrosine kinases (RTKs), as well as key components of the RAS‑RAF‑MEK‑ERK and PI3K/Akt/mTOR pathways. These drugs aim to turn down growth signals that cancer cells rely on, but their long‑term effectiveness is often limited by resistance and pathway rewiring.
RTK Inhibitors
Multiple targeted therapies act at the level of RTKs, blocking aberrant signals before they reach downstream pathways. Over a dozen EGFR and RTK inhibitors are now in use—from first-generation agents like gefitinib to newer, fourth-generation inhibitors such as taletrectinib. Among these, lorlatinib is a third-generation RTK inhibitor approved for ALK- and ROS1-positive cancers.
Signaling Molecule Inhibitors
Direct inhibitors of RAS-RAF-MEK-ERK and PI3K/AKT signaling molecules have also shown clinical benefit in some settings. Two KRAS G12C inhibitors, sotorasib and adagrasib, have been approved by the US Food and Drug Administration, along with multiple BRAF inhibitors (such as vemurafenib, dabrafenib, and encorafenib) and MEK inhibitors (including trametinib, binimetinib, selumetinib, and cobimetinib). PI3K inhibitors such as copanlisib, idelalisib, duvelisib, alpelisib, and umbralisib target different PI3K isoforms to dampen growth and survival signaling.
The mTOR pathway can be targeted using allosteric mTOR inhibitors such as rapamycin and its analogs, which have been available since the mid‑1990s, as well as newer ATP‑competitive mTOR kinase and complex inhibitors (for example, torin‑1 and torin‑2) that more broadly suppress mTORC1 and mTORC2 activity in preclinical models.
Future Directions & Preventing Therapy Resistance
Across all of these inhibitor classes, cancer cells can adapt by activating alternative receptors, engaging parallel pathways, or acquiring secondary resistance mutations, which limits the durability of responses. This has driven a shift toward combination and next‑generation approaches.
The number of second‑, third‑, and fourth‑generation RTK and Ras pathway inhibitors in preclinical and clinical testing continues to rise, including agents such as elironasib for KRAS G12C mutants and brigatinib for crizotinib‑resistant ALK‑driven cancers. Combining RAS‑targeted therapies with upstream RTK inhibitors, downstream SHP2, SOS1, or ERK inhibitors, or immune checkpoint inhibitors can improve outcomes compared with monotherapy.
Other promising strategies include targeted protein degradation, such as RTK degraders (for example, PROTACs) and bispecific degraders designed to overcome acquired resistance to kinase inhibitors. Recently, the AKT kinase inhibitor capivasertib was approved for combination therapy in HER2‑negative, hormone receptor–positive breast cancer with mutations in PIK3CA, AKT, or PTEN, after longer progression‑free survival was observed with dual therapy compared with hormone receptor–targeted therapy alone.
These advances highlight how challenging it is to fully shut off proliferative signaling, but also show how iterative drug design and rational combinations are paving the way for more effective and personalized treatments.
Additional Resources
Read the additional blogs in the Hallmarks of Cancer Series:
- Evading Growth Suppressors
- Nonmutational Epigenetic Reprogramming
- Avoiding Immune Destruction
- Tumor-Promoting Inflammation
- Activating Invasion & Metastasis
- Inducing or Accessing Vasculature (Angiogenesis)
- Senescent Cells
- Genome Instability & Mutation
- Resisting Cell Death
- Deregulating Cellular Metabolism
- Unlocking Phenotypic Plasticity
References:
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57-70. doi:10.1016/s0092-8674(00)81683-9
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-674. doi:10.1016/j.cell.2011.02.013
- Ciccarone F, Ciriolo MR. Editorial: Hallmark of cancer: sustained proliferative signalling. Front Oncol. 2023;13:1328827. Published 2023 Nov 23. doi:10.3389/fonc.2023.1328827
- Sever R, Brugge JS. Signal transduction in cancer. Cold Spring Harb Perspect Med. 2015;5(4):a006098. Published 2015 Apr 1. doi:10.1101/cshperspect.a006098
- Fujita N, Sato S, Katayama K, Tsuruo T. Akt-dependent phosphorylation of p27Kip1 promotes binding to 14-3-3 and cytoplasmic localization. J Biol Chem. 2002;277(32):28706-28713. doi:10.1074/jbc.M203668200
- Castellano E, Downward J. RAS Interaction with PI3K: More Than Just Another Effector Pathway. Genes Cancer. 2011;2(3):261-274. doi:10.1177/1947601911408079
- Hong D, Zhou B, Zhang B, et al. Recent advances in the development of EGFR degraders: PROTACs and LYTACs. Eur J Med Chem. 2022;239:114533. doi:10.1016/j.ejmech.2022.114533
- Oya Y, Imaizumi K, Mitsudomi T. The next-generation KRAS inhibitors…What comes after sotorasib and adagrasib?. Lung Cancer. 2024;194:107886. doi:10.1016/j.lungcan.2024.107886
- Turner NC, Oliveira M, Howell SJ, et al. Capivasertib in Hormone Receptor-Positive Advanced Breast Cancer. N Engl J Med. 2023;388(22):2058-2070. doi:10.1056/NEJMoa221413118-CEL-47850