Nonmutational epigenetic reprogramming is a defining characteristic of cancer, whereby tumor cells acquire heritable changes in gene expression without altering the underlying DNA sequence. Unlike permanent genetic alterations, epigenetic reprogramming occurs through a variety of transient yet inheritable mechanisms that are propagated as cancer cells divide.
The major mechanisms involved in nonmutational epigenetic reprogramming include DNA methylation, histone modification, chromatin remodeling, and regulation by non-coding RNAs (ncRNAs). This blog focuses on the proteins and enzymes responsible for DNA methylation and histone modification and the emerging therapeutic strategies that target the epigenetic landscape of the tumor microenvironment (TME).
< Jump to the product list at the end of this blog >
Cancer Epigenetics
|
Cancer cells hijack epigenetic mechanisms to alter gene expression in order to support their growth and survival. This is done through the activity of key enzymatic writer, reader, and eraser complexes, which extensively modify the chromatin during tumor progression. DNA methylation and histone modifications are dramatically altered in key oncogenes and tumor suppressors, enabling gene programs that assist in epithelial-to-mesenchymal transition (EMT), as well as adapting cells to nutrient deprivation, inflammation, and hypoxia from the TME. |
|
What are the Hallmarks of Cancer?The Hallmarks of Cancer1-3 are a research framework that organizes the key traits cancer cells acquire in order to grow and spread. Initially described by Douglas Hanahan and Robert Weinberg in 2000, the framework groups the underlying mechanisms of cancer into a series of smaller subsets to advance discovery. The concept was expanded in 2011 to include two additional hallmarks and two enabling characteristics, and again in 2022 with four new emerging hallmarks.
|
 Identified in 2011, Nonmutational Epigenetic Reprogramming is one of the expanded core cancer hallmarks.
Identified in 2011, Nonmutational Epigenetic Reprogramming is one of the expanded core cancer hallmarks.These heritable changes are very similar to those found during stem cell reprogramming and involve many of the same enzymes.
DNA Methylation in Cancer
Abnormal DNA methylation is one of the most common epigenetic changes in human cancers.4 This includes reduced global DNA methylation or hypomethylation, which contributes to tumorigenesis by activating oncogenes and inducing chromosomal instability. Conversely, cancer is also often associated with the hypermethylation of CpG islands at gene promoters of tumor suppressors, leading to reduced expression.
 |
DNA methylation is a key epigenetic modification implicated in tumor development. Download the DNA Methylation Pathway diagram, along with a list of relevant CST products. |
These changes are orchestrated by DNA writers, readers, and erasers, enzymes that apply, interpret, and remove methylation marks to regulate gene expression. A disruption in the balance of these enzymes leads to the aberrant methylation patterns seen in many types of cancer.
Writers: DNA Methyltransferase (DNMT) Family Proteins
Writers are enzymes that apply epigenetic marks, such as methyl groups, to specific targets. The DNA methyltransferase (DNMT) family, responsible for DNA methylation, includes the de novo methyltransferases DNMT3A and DNMT3B, which are essential for mammalian development, and the maintenance methyltransferase, DNMT1, which methylates newly synthesized DNA during cellular replication. Aberrant expression of DNMT family members is associated with many forms of cancer.5
DNA methyltransferases are particularly significant in hematological malignancies. For example, DNMT3A mutations are frequently found in acute myeloid leukemia (AML), where they contribute to altered methylation patterns. Similarly, abnormal DNMT3A and DNMT1 activity has been implicated in T-cell acute lymphoblastic leukemia (T-ALL), where it affects gene expression programs critical for normal T-cell differentiation and function.
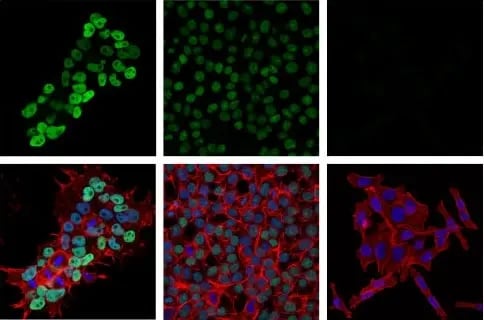
Immunofluorescent (IF) analysis of NCCIT embryonal carcinoma cells (left, DNMT3B high-expressing), HCT 116 DNMT3B wild-type colorectal carcinoma cells (middle, DNMT3B low-expressing), and HCT 116 DNMT3B knockout cells (right, DNMT3B negative) using DNMT3B (E8A8A) Rabbit Monoclonal Antibody #57868 (green). Actin filaments were labeled with DyLight® 554 Phalloidin #13054 (red). Samples were mounted in ProLong Gold Antifade Reagent with DAPI #8961 (blue).
Therapeutically, first-generation DNMT inhibitors (DNMTis), including cytosine analogues 5-azacytidine (AZA) and decitabine, were approved by the US Food and Drug Administration (FDA) in 2004 and 2006 for the treatment of myelodysplastic syndrome (MDS). These agents induce DNA hypomethylation and the reactivation of silenced tumor suppressor genes. However, they are also associated with significant toxicity and have short half-lives, limiting their clinical effectiveness.
Second-generation DNMTis, such as SGI-110 (guadecitabine) and MG98, were developed to improve efficacy and stability, but ultimately failed to achieve FDA approval. More recently, the DNMT1-specific inhibitor GSK3285032 has shown promise in research and preclinical studies focused on hematological malignancies.
In addition to direct enzymatic inhibition, researchers are now targeting DNMT-protein interactions, developing novel small molecules, and exploring combination therapies that pair DNMT inhibition with immune checkpoint blockade or other targeted agents. These approaches aim to enhance therapeutic windows, reduce toxicity, and activate antitumor immune responses—a growing area of focus within cancer epigenetics.
Erasers: TET Enzymes
DNA erasers remove epigenetic marks. Ten-eleven translocation (TET) enzymes, including TET1 and TET2, mediate DNA demethylation by converting 5-methylcytosine (5mC) into its oxidized forms—hydroxymethylcytosine (5hmC), formylcytosine (5fC), and carboxylcytosine (5caC). The final step involves the excision of 5caC via base-excision repair, which effectively reverses DNA methylation.
TET1 and TET2 are frequently mutated or suppressed in cancers such as AML and lymphomas, particularly under hypoxic conditions, resulting in altered DNA methylation patterns and tumor promotion.6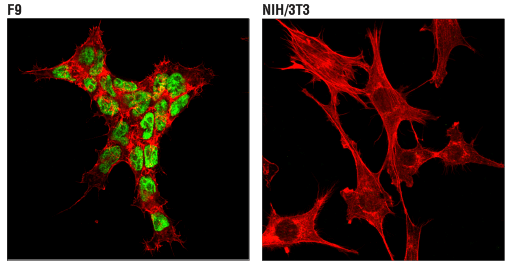
IF analysis of F9 mouse embryonal carcinoma cells (left, positive) and NIH/3T3 NIH/3T3 mouse fibroblasts (right, negative) using TET2 (D6C7K) Rabbit Monoclonal Antibody #36449 (green). Actin filaments were labeled with DyLight® 554 Phalloidin #13054 (red).
Imprinting Control Regions
Imprinting control regions (ICRs) regulate specific groups of genes critical for normal development. The CCCTC-binding factor (CTCF), which maintains topologically associating domains (TADs), is highly susceptible to altered DNA methylation that can disrupt these domains, juxtaposing oncogenes with constitutively active enhancers and leading to the amplification of malignant signaling.
Histone Modifications in Cancer
In addition to DNA methylation, histone modifications alter chromatin accessibility and can activate oncogenes or inactivate tumor-suppressor genes. The following are some of the key histone modifiers being targeted to treat cancer.
 |
Download the Epigenetic Writers and Erasers of Histone H3 diagram for a list of the writers and eraser proteins, along with relevant CST antibodies. |
 |
Download the Epigenetic Writers and Erasers of Histones H2A, H2B, and H4 diagram for a list of the writers and eraser proteins, along with relevant CST antibodies. |
Writers: Polycomb Repressive Complex 2 (PRC2)
The Polycomb Repressive Complex 2 (PRC2) is responsible for the trimethylation of histone H3 at lysine 27 (H3K27me3), an epigenetic mark that silences gene expression and is mediated by its catalytic subunit, Enhancer of zeste homolog 2 (Ezh2). PRC2 and Ezh2 activity is regulated by multiple transcription factors that are aberrantly regulated in cancer, including Myc, E2F/Rb, Elk-1, and HIF-1. The overexpression of Ezh2 in various cancers can contribute to epithelial-mesenchymal transition (EMT), tumor growth, and metastasis.
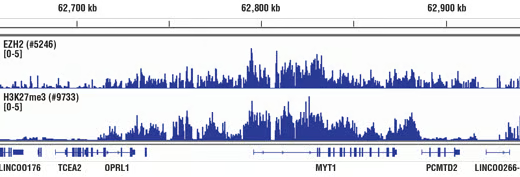 |
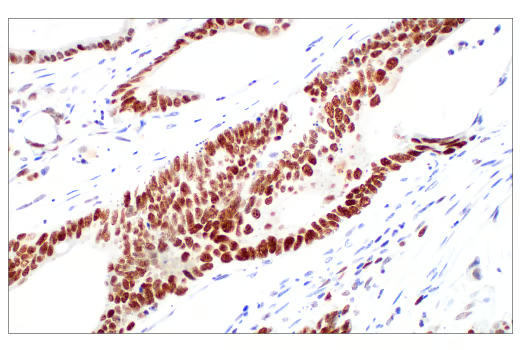 |
| ChIP performed with cross-linked chromatin from HeLa cells and either Ezh2 (D2C9) Rabbit Monoclonal Antibody #5246 or Tri-Methyl-Histone H3 (Lys27) (C36B11) Rabbit Monoclonal Antibody #9733, using SimpleChIP® Enzymatic Chromatin IP Kit (Magnetic Beads) #9003. DNA Libraries were prepared using DNA Library Prep Kit for Illumina (ChIP-seq, CUT&RUN) #56795. EZH2 and H3K27me3 are known to associate with each other on chromatin. The figure shows binding of both EZH2 and H3K27me3 across the MYT1 gene. | Immunohistochemical (IHC) analysis of paraffin-embedded human colon adenocarcinoma using Ezh2 (D2C9) XP Rabbit Monoclonal Antibody #5246 performed on the Leica BOND Rx. Ezh2 is overexpressed in many cancers. |
Ezh2 catalyzes histone methylation by transferring methyl groups from the methyl donor S-adenosylmethionine (SAM) to histone H3, producing S-adenosyl-L-homocysteine (SAH) as a byproduct, which can inhibit methyltransferase activity if it accumulates.7
Targeting Ezh2 has emerged as a promising strategy to reverse epigenetic gene silencing in cancer. Ezh2 inhibitors either block the catalytic activity of Ezh2—often by binding directly to the SAM-binding pocket—or affect the structure of the PRC2 complex. This inhibits the methyltransferase activity of Ezh2, leading to the reactivation of tumor suppressor genes.
Clinically, the first-in-class Ezh2 inhibitor tazemetostat was approved by the US FDA in 2020 for the treatment of refractory follicular lymphoma and epithelioid sarcoma, marking a significant advance in targeting PRC2 in cancer therapy. Multiple ongoing clinical trials are currently evaluating tazemetostat as both a monotherapy and in combination with other treatments, with the goal of expanding its use to additional cancer types.
Recent studies have also found that PRC2 can also act as a reader of histone marks, allowing for the dynamic recognition of methylated chromatin to reinforce repressive domains.8
Erasers: Histone Deacetylases (HDACs)
Histone deacetylases (HDACs) catalyze the removal of acetyl groups from histones to regulate chromatin structure and gene transcription. They are differentially expressed in tumors and contribute to oncogenesis through mechanisms including the following:
- HDACs can upregulate survival signatures by removing acetyl groups from key apoptotic genes such as p53, BAX, and PUMA.
- HDAC1 and HDAC2 bind p27 and p21, affecting cell cycle and G1/S transition.8
- HDAC1 and HDAC2 also provide resistance to DNA damage by modifying chromatin structure around double-stranded breaks and interacting with DNA repair machinery.9
- SirT1 deacetylates the DNA repair factor Ku70, causing it to sequester the proapoptotic factor Bax and inhibiting stress-induced apoptotic cell death.10

- SirT1 also deacetylates hypoxia-inducible factor 1 alpha (HIF-1α), thereby inactivating it to repress the expression of HIF1 target genes.11
Several HDAC inhibitors have been approved for use by the US Food and Drug Administration, including Vorinostat in 2006, and Romidepsin in 2009, for the treatment of cutaneous T-cell lymphoma (CTCL). Belinostat was approved in 2013 to treat refractory peripheral T-cell lymphoma (PTCL). HDAC inhibitors work by blocking the catalytic activity of HDACs to cause increased acetylation, which can lead to reactivation of pro-apoptotic genes and cause growth arrest and cell death.
Readers: Bromodomain and Extraterminal Domain (BET) Family Proteins
The BET family of proteins, consisting of BRD1, BRD2, BRD3, BRD4, and BRDT, contributes to cancer development by binding to acetylated histones over oncogene promoters such as c-Myc. Specifically, the bromodomain itself forms a hydrophobic pocket that captures acetylation modifications. BRD4 is overexpressed in many different tumor types, where it also controls other oncogenic proteins such as Myb, ERG, E2F1k, and NF-kB. The BRD4-NUT1 translocation mutation has been widely studied for its role in NUT midline carcinoma (NMC), a highly aggressive subtype of squamous cell cancer.
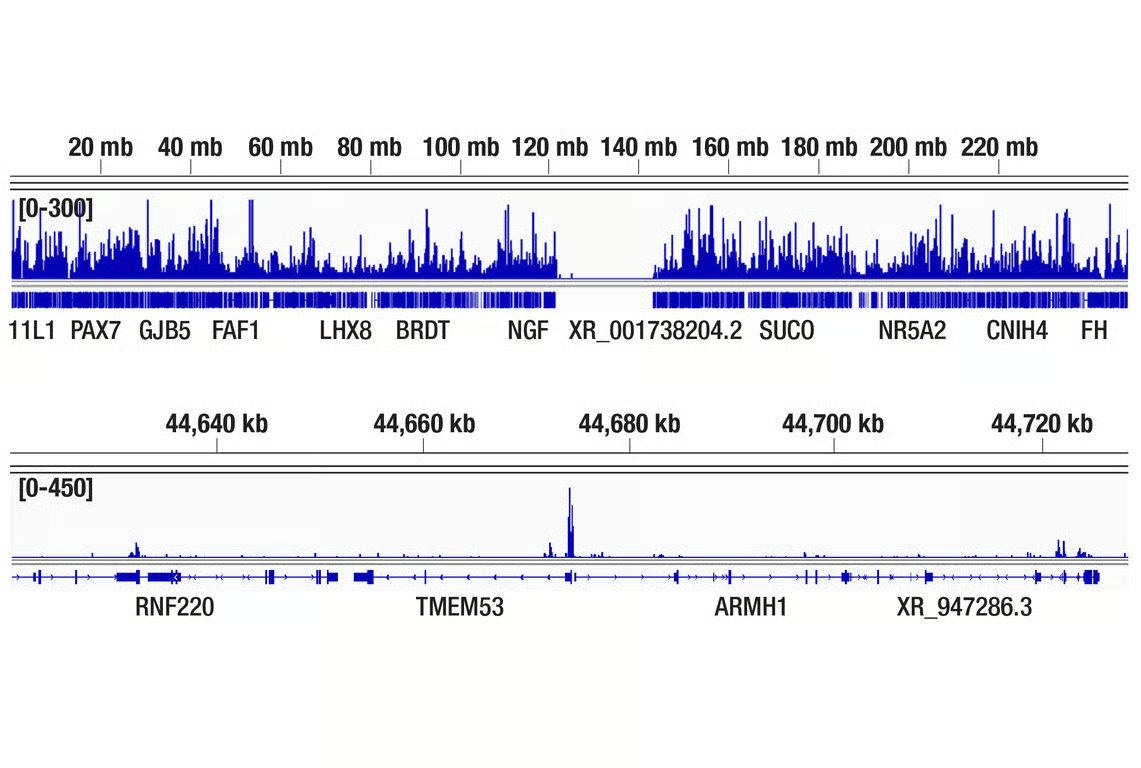
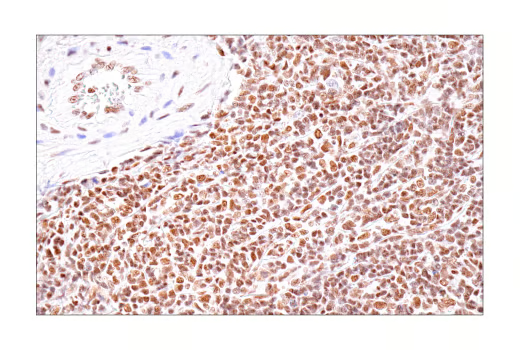
The discovery of BET inhibitors such as JQ1, a molecule that selectively binds the BET bromodomain of BRD2/3/4 to disrupt interactions with acetylated histones, represents an important breakthrough for cancer research. JQ1 has been widely used in preclinical studies to explore epigenetic regulation and mechanisms in various cancers, including multiple myeloma, where it can lower Myc activity. However, JQ1 itself is not FDA approved for any indication, largely due to its short half-life and poor pharmacokinetic properties; it serves as a chemical probe and tool compound rather than a therapeutic. Research is ongoing to develop modified BET inhibitors, including pan-BET inhibitors, bivalent ligands, and PROTACs, but as of 2025, none of these classes have received FDA approval for cancer treatment.
Rewriting Cancer Epigenetics
The epigenetic signature established during oncogenesis is a powerful way in which cancer cells harness existing cellular machinery to acquire the traits necessary to grow and spread. Histone modifications and DNA methylation are drastically different in cancer cells versus normal cells, resulting in differential expression of key oncogenic transcription factors.
Unlike genetic mutations, however, epigenetic marks can be reversed, offering opportunities for therapeutic intervention. Treatments that reset or disrupt the cancer epigenome, especially when used alongside a mainline therapy, are a promising strategy for effectively targeting cancer.
| Chromatin Profiling Kits |
| CUT&RUN Assay Kit #86652 |
| CUT&Tag Assay Kit #77552 |
| SimpleChIP® Enzymatic Chromatin IP Kit (Magnetic Beads) #9003 |
| SimpleChIP® Plus Sonication Chromatin IP Kit #56383 |
Additional Resources
Read the additional blogs in the Hallmarks of Cancer Series:
- Evading Growth Suppressors
- Avoiding Immune Destruction
- Tumor-Promoting Inflammation
- Activating Invasion & Metastasis
- Inducing or Accessing Vasculature (Angiogenesis)
- Senescent Cells
- Genome Instability & Mutation
- Resisting Cell Death
- Deregulating Cellular Metabolism
- Unlocking Phenotypic Plasticity
- Sustaining Proliferative Signaling
References
- Hanahan D, Weinberg RA (January 2000). "The Hallmarks of Cancer". Cell. 100 (1): 57–70. doi:10.1016/S0092-8674(00)81683-9
- Hanahan D, Weinberg RA (March 2011). "Hallmarks of Cancer: the next generation". Cell. 144 (5):646-74. doi: 10.1016/j.cell.2011.02.013.
- Hanahan D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022;12(1):31-46. doi:10.1158/2159-8290.CD-21-1059
- Kanai Y, Hirohashi S. Alterations of DNA methylation associated with abnormalities of DNA methyltransferases in human cancers during transition from a precancerous to a malignant state. Carcinogenesis. 2007;28(12):2434-2442. doi:10.1093/carcin/bgm206
- Jin B, Robertson KD. DNA methyltransferases, DNA damage repair, and cancer. Adv Exp Med Biol. 2013;754:3-29. doi:10.1007/978-1-4419-9967-2_1
- Thienpont B, Van Dyck L, Lambrechts D. Tumors smother their epigenome. Mol Cell Oncol. 2016;3(6):e1240549. doi:10.1080/23723556.2016.1240549
- Parreno V, Loubiere V, Schuettengruber B, et al. Transient loss of Polycomb components induces an epigenetic cancer fate. Nature. 2024;629(8012):688-696. doi:10.1038/s41586-024-07328-w
- Uckelmann M, Davidovich C. Not just a writer: PRC2 as a chromatin reader. Biochem Soc Trans. 2021;49(3):1159-1170. doi:10.1042/BST20200728
- Tiwari N, Tiwari VK, Waldmeier L, et al. Sox4 is a master regulator of epithelial-mesenchymal transition by controlling Ezh2 expression and epigenetic reprogramming. Cancer Cell. 2013;23(6):768-783. doi:10.1016/j.ccr.2013.04.020
- Shah RR. Safety and Tolerability of Histone Deacetylase (HDAC) Inhibitors in Oncology. Drug Saf. 2019;42(2):235-245. doi:10.1007/s40264-018-0773-9
- Miller KM, Tjeertes JV, Coates J, et al. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat Struct Mol Biol. 2010;17(9):1144-1151. doi:10.1038/nsmb.1899
- Cohen HY, Miller C, Bitterman KJ, et al. Calorie Restriction Promotes Mammalian Cell Survival by Inducing the SIRT1 Deacetylase. Science. 305,390-392(2004).DOI:10.1126/science.1099196
- Lim JH, Lee YM, Chun YS, Chen J, Kim JE, Park JW. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1 alpha. Mol Cell. 2010;38(6):864-878. doi:10.1016/j.molcel.2010.05.023