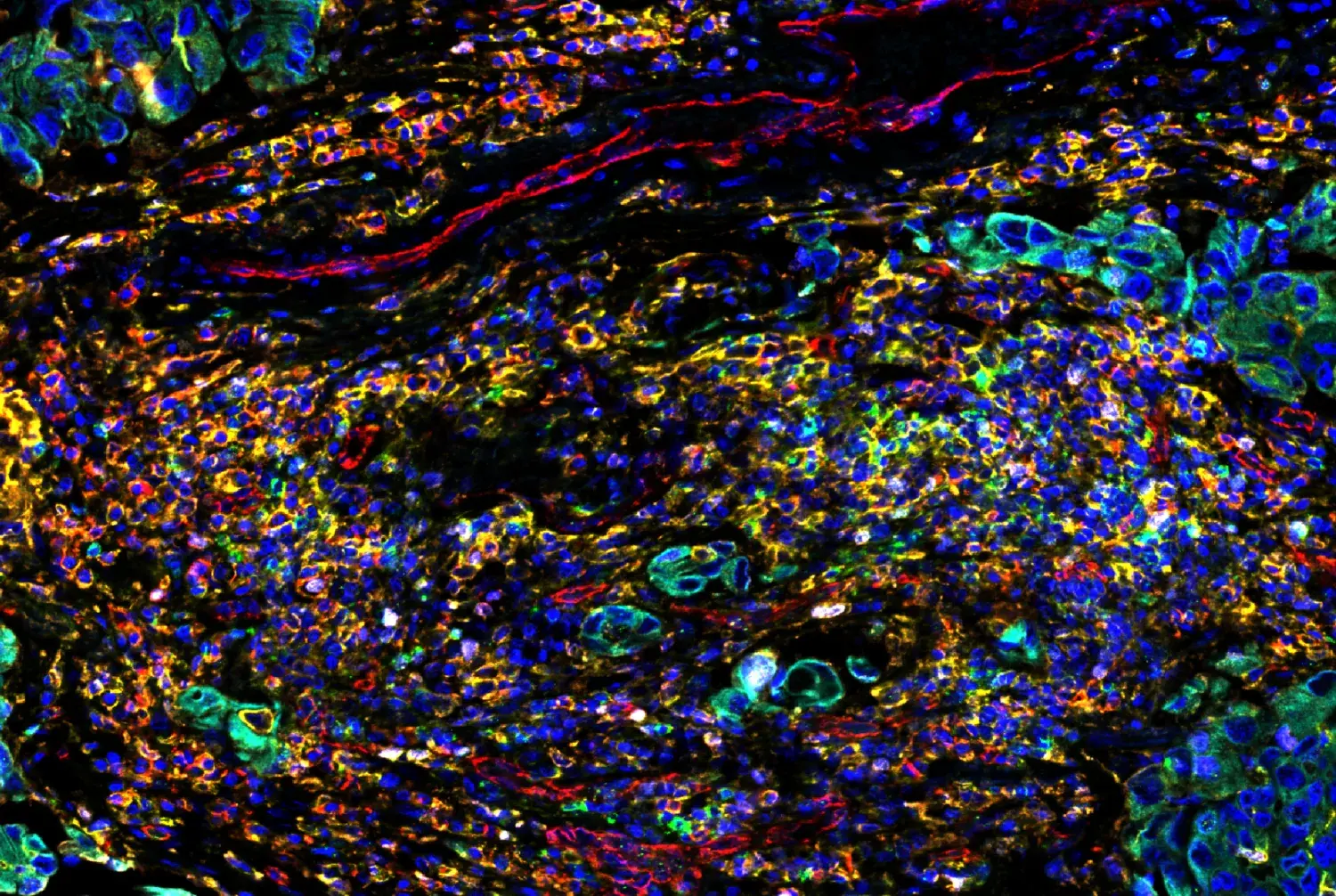
Imagine a rapidly growing city—it needs roads, plumbing, and power lines to thrive. So too, do tumors. To grow beyond a tiny cluster of cells, tumors need their own supply lines for oxygen and nutrients, a means to evacuate waste products and CO2, and highways to spread to distant sites. This vital network is formed through angiogenesis, a process during which new blood vessels are generated from preexisting ones.
This blog explores some of the signaling mechanisms and events that enable tumors to induce angiogenesis, one of the Hallmarks of Cancer, many of which are being targeted for therapeutic intervention.
< Jump to the product list at the end of this blog >

Developmental Angiogenesis vs Tumor Vasculature
Under normal physiological conditions, angiogenesis is tightly regulated by signaling pathways that either promote or block the growth of new blood vessels. In vertebrates, most of the vascular system is laid out during embryonic development in two distinct and sequential processes: vasculogenesis and angiogenesis.
|
In the developing embryo, vasculogenesis is the process through which mesenchymal stem cells differentiate into endothelial cells, and a primitive vascular system is laid out. Once the initial framework is established, angiogenesis takes over, expanding the network by sprouting new vessels from existing ones. The process is tightly regulated and coordinated to ensure the formation of a functional and stable vascular system. Once developed, the system remains largely stable through adulthood, only becoming transiently reactivated during wound repair and menstruation, for example. |
|
What are the Hallmarks of Cancer?The Hallmarks of Cancer1-3 are a landmark conceptual framework that organizes the key traits cancer cells acquire in order to grow, survive, and spread. Initially described by Douglas Hanahan and Robert Weinberg in 2000, the framework distills the underlying mechanisms of cancer into a series of smaller subsets to advance discovery. Initially comprising six core hallmarks, the concept was expanded in 2011 to include two additional hallmarks and two enabling characteristics, and again in 2022 with one new hallmark and four new enabling characteristics.
|
 Inducing or Accessing Vasculature is one of the original hallmarks, first described as Sustained Angiogenesis in the seminal Hallmarks of Cancer (2000).
Inducing or Accessing Vasculature is one of the original hallmarks, first described as Sustained Angiogenesis in the seminal Hallmarks of Cancer (2000).The well-organized vascular architecture is not unlike the main roads and blocks being laid out in a new, well-planned city.
In contrast, tumor angiogenesis is a pathological process in which some of these same mechanisms are co-opted and dysregulated. The re-wiring of the tumor vasculature is driven largely by the sprouting mechanism via pro-angiogenic factors, most notably vascular endothelial growth factor (VEGF) signaling. A tumor's rapid and uncontrolled growth results in a chaotic, leaky, and disorganized vascular network that is poorly perfused. If healthy vasculature resembles a well-planned neighborhood, tumor angiogenesis is a sprawling city with poorly planned roads, unstable bridges, and leaky sewers. This not only creates an immunosuppressive environment that is difficult for anti-tumor immune cells to infiltrate and patrol, it can also enable escape routes for rogue cancer cells to travel to distant organs and seed secondary tumors (metastases).
Angiogenesis & Tumor Angiogenesis Signaling Pathways
 |
Download the Angiogenesis Signaling Pathway diagram along with relevant CST products.
|
 |
Explore the Tumor Angiogenesis Signaling Pathway diagram along with relevant CST products. |
Features of Tumor Vasculature
Tumor vessels are frequently dilated, uneven, and saccular, often forming trifurcations instead of the hierarchical bifurcated branching found in normal vasculature. The endothelial cell lining (marker: CD31/PECAM-1) is structurally compromised, with discontinuous basement membranes and incomplete cell junctions due to reduced VE-cadherin expression. This causes blood vessels to become notoriously leaky, leading to vascular permeability and fluid extravasation.
As a result, blood flow within a tumor is often disorganized and heterogeneous, with regions of stasis or shunting, the latter referring to places where blood vessels in or around a tumor bypass capillary-rich tissue to take a shorter, more direct route. Additionally, newly formed tumor vessels often lack adequate coverage by pericytes (marker: α-smooth muscle actin)—a type of mural cell that normally provides structural support to capillaries—making them fragile and prone to collapse or regression.
 SignalStar® Multiplex IHC staining of paraffin-embedded human gastric adenocarcinoma performed on the Leica BOND RX using the following: CD31 (PECAM-1) (89C2) & CO-0028-488 SignalStar Oligo-Antibody Pair #83823 (green), CD68 (D4B9C) & CO-0007-594 SignalStar Oligo-Antibody Pair #77318 (yellow), Pan-Keratin (C11) & CO-0003-647 SignalStar Oligo-Antibody Pair #16373 (red), α-Smooth Muscle Actin (D4K9N) & CO-0024-750 SignalStar Oligo-Antibody Pair #29280 (cyan), and ProLong Gold Antifade Reagent with DAPI #8961 (blue).
SignalStar® Multiplex IHC staining of paraffin-embedded human gastric adenocarcinoma performed on the Leica BOND RX using the following: CD31 (PECAM-1) (89C2) & CO-0028-488 SignalStar Oligo-Antibody Pair #83823 (green), CD68 (D4B9C) & CO-0007-594 SignalStar Oligo-Antibody Pair #77318 (yellow), Pan-Keratin (C11) & CO-0003-647 SignalStar Oligo-Antibody Pair #16373 (red), α-Smooth Muscle Actin (D4K9N) & CO-0024-750 SignalStar Oligo-Antibody Pair #29280 (cyan), and ProLong Gold Antifade Reagent with DAPI #8961 (blue).
The combination of leaky vasculature and lack of functional lymphatic drainage increases interstitial fluid pressure (IFP), which acts as a physical barrier to the delivery of oxygen, immune cells, chemotherapeutic drugs, and other therapeutic agents. As a result, tumors become increasingly acidic and hypoxic (low oxygen levels), leading to resistance to treatments such as radiation and chemotherapy.
Hypoxia, Tumorigenesis, and the “Angiogenic Switch”
One of the earliest events in tumorigenesis, when tumor masses are still microscopic, is the tripping of the “angiogenic switch,” which occurs when the balance between pro- and anti-angiogenic factors tips in favor of new blood vessel formation. The most potent pro-angiogenic signaling pathway driving this process is induced by hypoxia and is mediated by members of the Hypoxia-Inducible Factor-1 (HIF-1) and VEGF protein families.

In normal, well-oxygenated cells, HIF-1α is hydroxylated by oxygen-sensing prolyl-4-hydroxylases (PHDs), which marks it for degradation and prevents its buildup. However, under the hypoxic conditions of the TME, HIF-1α escapes degradation, translocates to the nucleus, and joins with HIF-1β to form a transcription factor complex. This complex then “switches on” genes to up-regulate the expression of numerous proteins involved in angiogenesis, most notably VEGF-A.
 |
 |
| IHC analysis showing nuclear localization of HIF-1α in paraffin-embedded gastrointestinal stromal tumor using HIF-1α (E1V6A) Rabbit Monoclonal Antibody #48085. |
Cobalt chloride is a widely used cell treatment that mimics the effects of hypoxia on HIF1A proteins, leading to their increased stability and subsequent translocation to the nucleus. Immunofluorescent (IF) analysis of HepG2 cells (human liver cancer), untreated (left) or treated with cobalt chloride, using HIF-1α (D1S7W) Rabbit Monoclonal Antibody #36169 (green). |
It’s worth noting that tumor-associated macrophages (TAMs) within the TME also produce VEGF-A, in addition to other pro-angiogenic mediators. This can further amplify the angiogenic switch to facilitate tumor vascularization.
Tumor Angiogenesis Signaling Pathways & Proteins
Once the angiogenesis switch is flipped in tumor cells, pro-angiogenic signaling cascades are activated. To induce vasculature, VEGF-A binds to its primary receptor, VEGF Receptor 2 (KDR/Flk-1), reawakening quiescent endothelial cells in existing blood vessels. This results in receptor phosphorylation and dimerization, and initiates signaling through the PI3K/Akt, mTOR, and MAPK/ERK pathways to promote endothelial cell proliferation and migration.
 |
 |
|
IHC analysis of paraffin-embedded human angiosarcoma using VEGF Receptor 2 (D5B1) Rabbit Monoclonal Antibody #9698.
|
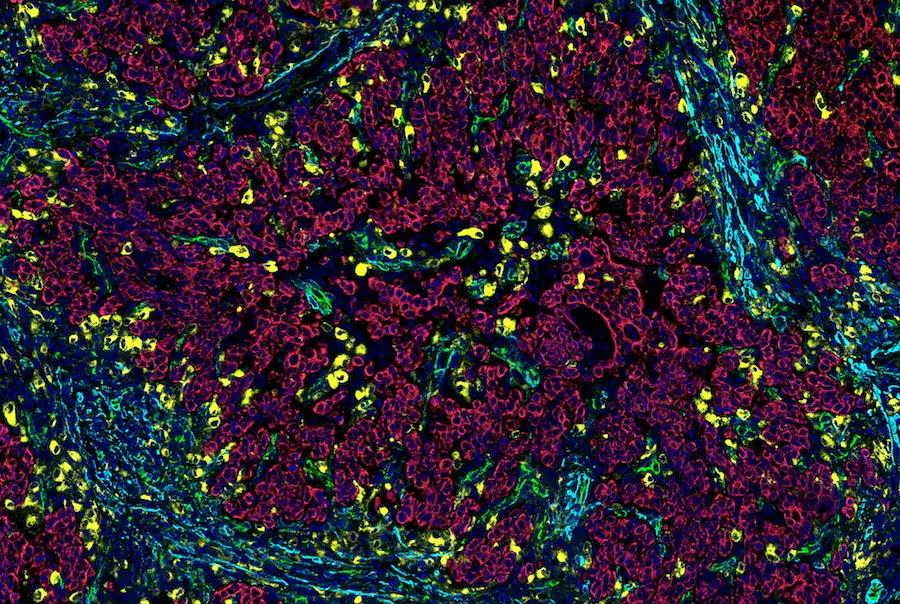
VEGFR2 can be detected on the cell surface of endothelial cells in many tumors. Left: IHC analysis of paraffin-embedded breast angiosarcoma using VEGF Receptor 2 (55B11) Rabbit Monoclonal Antibody #2479. Right: A serial section is stained with the endothelial cell marker CD31 (PECAM-1). |
Activated endothelial tip cells at the leading edge of the angiogenic sprout secrete matrix metalloproteases (MMPs) that break down the surrounding extracellular matrix (ECM), including the basement membranes of existing vasculature. This creates a path for the tip cells to migrate into the tumor, facilitated by integrins such as αv β3 and α5 β1, which enable the adhesion of the endothelial tip cells to the provisional ECM. Behind the tip cells, stalk cells proliferate, lengthening the new vessel sprout. Ultimately, newly formed sprouts connect to form lumens and establish a functional vascular network around the tumor.
In parallel to this process, PDGF-β is secreted by activated endothelial cells and binds to its receptor, PDGFR-β, on pericytes. This guides the recruitment of pericytes to the growing spout, where they help stabilize the growing vascular system.
Many other ligands and signaling pathways are also involved in tumor angiogenesis. These include the following:

|
DLL4/Notch Signaling PathwayThe DLL4/Notch signaling pathway helps regulate the balance between tip cells and stalk cells to ensure organized sprouting. Normally, migratory tip cells have low Notch levels, while proliferative stalk cells exhibit high Notch expression. In tumors, dysregulated DLL4/Notch signaling can lead to chaotic vessel formation and increased vascular permeability, and contributes to tumor metastasis. View the Notch Signaling Pathway |

|
Angiopoietin and Tie Receptor SignalingThe angiopoietin family (Ang1, Ang2) and their receptors (Tie1, Tie2) regulate blood vessel maturation and stability. In normal tissues, Ang1/Tie2 signaling supports endothelial cell survival, strengthens endothelial cell–pericyte interactions, and maintains mature blood vessels in a non-sprouting state. Ang2 expression is normally low and restricted to sites of vascular remodeling (e.g., the female reproductive tract), where it acts as a natural Ang1 antagonist to promote vessel destabilization when needed. In many cancers, however, Ang2 is dramatically upregulated and correlates with tumor malignancy. Its function is context-dependent: In the absence of VEGF-A, Ang2 acts as a Tie2 agonist, disrupting endothelial cell-pericyte connections and promoting vessel regression, leading to increased hypoxia. In response to the high VEGF-A levels found in tumors, Ang2 instead induces endothelial activation and sprouting by destabilizing quiescent endothelial cells and promoting their proliferation and migration. Increased Ang2 levels can also inhibit the stabilizing effects of Tie2, shifting cellular balance toward dysfunctional vessels. It can also directly stimulate endothelial cell proliferation and migration, and contributes to the characteristic leakiness of tumor vessels. View the Tumor Angiogenesis Signaling Pathway |

|
Transforming Growth Factor-β (TGF-β) SignalingMany tumors have high tissue levels of TGF-β, which is known to induce vessel sprouting and upregulate the matrix metalloproteases MMP2 and MMP9, which act on the extracellular matrix (ECM) to promote angiogenesis and tumor invasiveness. TGF-β also increases expression of CTGF, VEGF, bFGF, and IL-1, which further contribute to angiogenesis. Beyond angiogenesis, TGF-β also contributes to epithelial-to-mesenchymal transition (EMT), invasion, fibrosis, and suppression of anti-tumor immunity. View the TGF-β Signaling Pathway |
Other VEGF receptors
While VEGF-A mainly drives angiogenesis via its interaction with VEGF Receptor 2, it also binds to VEGF Receptor 1 (Flt-1), which regulates vessel sprouting and myeloid cell recruitment. VEGF-C and VEGF-D bind to receptor VEGFR3 (Flt-4) and are primarily involved in promoting lymphangiogenesis (lymphatic vessel growth). Fibroblast Growth Factors (FGFs) also promote angiogenesis, often acting synergistically with VEGF signaling pathways.
Additionally, the NFκB signaling pathway plays a role in tumor angiogenesis by upregulating a host of pro-angiogenic and pro-survival genes, including VEGF-A and Interleukin-8 (IL-8), which drive proliferation and migration of nearby endothelial cells. NFκB also promotes the transcription of Matrix Metalloproteinases (MMPs), such as MMP-2 and MMP-9. These enzymes are essential for breaking down the extracellular matrix (ECM), which is a prerequisite for both endothelial cell sprouting and tumor cell invasion. In the tumor cells, NFκB activity promotes cell survival and proliferation, which is foundational to tumor growth and its subsequent need for a new blood supply. This activity of NFκB is one of the major reasons why tumors can sustain an inflammatory and vascularized microenvironment.
Immune Component and Interplay with the Tumor Microenvironment
The tumor vasculature is not merely a supply line. It also serves as an active and influential component of the tumor microenvironment, interacting closely with immune cells and other stromal elements to drive cancer progression.
Immune Cell Contribution to Angiogenesis
- Tumor-Associated Macrophages (TAMs): TAMs are key players in tumor development, often polarized to a pro-tumorigenic M2-like phenotype. They secrete a plethora of pro-angiogenic factors, including VEGF, MMPs, and various cytokines (e.g., IL-8, TNF-α, and HGF), further fueling angiogenesis.
- Neutrophils: These can promote tumor angiogenesis by releasing MMPs and triggering the release of VEGF and other angiogenic factors.
- Other Immune Cells: Lymphocytes (e.g., B cells and T cells) can indirectly influence angiogenesis through the secretion of various pro-angiogenic mediators.
- Stromal Cells: In addition to immune cells, adipocytes can promote angiogenesis within the tumor microenvironment (TME) by releasing a variety of pro-angiogenic factors. This effect is pronounced in fatty tissues or obesity, where adipocytes can become immunologically active and secrete immune signaling molecules that influence the TME.
Impact on Immune Cell Infiltration and Function
- Physical Barrier: The abnormal, leaky tumor vasculature, along with the dense ECM and high interstitial fluid pressure (IFP), creates a physical barrier that hinders the infiltration of anti-tumor immune cells (e.g., cytotoxic T lymphocytes) into the tumor core.
- Immunosuppressive Environment: The hypoxic and acidic TME, coupled with factors secreted by tumor cells and pro-tumorigenic immune cells (e.g., TAMs), creates an immunosuppressive milieu that impairs the function of infiltrating immune cells and promotes immune evasion.
- Vascular Endothelial Cells as Immunomodulators: Tumor endothelial cells (TECs) themselves can actively contribute to immunosuppression by expressing immune checkpoints or secreting factors that suppress anti-tumor immunity.
Blog: Hallmarks of Cancer: Avoiding Immune Destruction
Anti-Angiogenic Cancer Therapies
By understanding the intricate mechanisms of tumor vascularization, researchers have developed anti-angiogenic drugs, such as bevacizumab (Avastin), a monoclonal antibody that targets VEGF-A, and various small-molecule tyrosine kinase inhibitors (TKIs) that target VEGFRs (e.g., Sunitinib, Sorafenib, Pazopanib). These therapies aim to "normalize" tumor vessels or starve the tumor by inhibiting new vessel growth, improving the delivery of chemotherapy, and even enhancing immune responses.
However, while initial anti-angiogenic therapies showed promise, tumors often develop resistance. Current research is therefore focused on:
- Identifying new, less redundant angiogenic targets.
- Developing combination therapies (e.g., anti-angiogenics with immunotherapies or chemotherapy).
- Understanding the mechanisms of resistance.
- Developing therapies that normalize abnormal tumor vessels rather than just inhibiting their growth, to improve drug delivery and immune cell infiltration.
Unraveling the complexities of tumor vascularization remains a critical battleground in the fight against cancer, with ongoing research aimed toward uncovering new vulnerabilities in a tumor's vital supply lines.
| Product Name / Target | Applications | Reactivity |
| VEGF Signaling Pathway – The Master Regulator Most Important Targets: VEGF-A, VEGFR1, VEGFR2 (total & phosphorylated forms) |
||
| VEGF-A (E9X8Q) Rabbit mAb #50661 | WB, IP | H |
| VEGF-B Antibody #2463 | WB | H |
| VEGF-C Antibody #2445 | WB | H |
| VEGF Receptor 1 (E7T9H) Rabbit mAb #64094 | WB | H |
| VEGF Receptor 2 (D5B1) Rabbit mAb #9698 | WB, IP, IHC, IF, F | H, M, R |
| Phospho-VEGF Receptor 2 (Tyr951) (15D2) Rabbit mAb #4991 | WB | H, M |
| Phospho-VEGF Receptor 2 (Tyr996) Antibody #2474 | WB | H, M |
| Phospho-VEGF Receptor 2 (Tyr1059) (D5A6) Rabbit mAb #3817 | WB | H, M |
| Phospho-VEGF Receptor 2 (Tyr1175) (19A10) Rabbit mAb #2478 | WB | H, M |
| Phospho-VEGF Receptor 2 (Tyr1175) (D5B11) Rabbit mAb #3770 | WB, IP | H, M |
| VEGF Receptor 3 (D1J9Z) Rabbit mAb #33566 | WB, IP | H |
| Hypoxia-Inducible Factors (HIFs) – The Oxygen Sensors Most Important Target: HIF-1α |
||
| HIF-1α (D1S7W) Rabbit mAb #36169 | WB, IP, IF, F, ChIP, C&R | H, M, Mk |
| HIF-1α (D2U3T) Rabbit mAb #14179 | WB, ChIP, C&R | H, M, R, Mk |
| HIF-1α (E1V6A) Rabbit mAb #48085 | WB, IHC, IF | H, M |
| Hydroxy-HIF-1α (Pro564) (D43B5) Rabbit mAb #3434 | WB, IP | H, Mk |
| Fibroblast Growth Factor (FGF) Pathway Most Important Target: FGF2 (Basic FGF / bFGF) |
||
| Basic FGF (E5Y6M) Rabbit mAb #46879 | WB, IHC, IF | H, M |
| FGF Receptor 1 (FGFR1) (D8E4) Rabbit mAb #9740 | WB, IP, IHC, IF, F | H, M, Mk |
| Phospho-FGF Receptor 1 (Tyr653/654) (D4X3D) Rabbit mAb #52928 | WB, IP, IF | H |
| Platelet-Derived Growth Factor (PDGF) Pathway – Pericyte Recruitment Most Important Targets: PDGFR β (total & phosphorylated forms), α-SMA (as a pericyte/smooth muscle marker) |
||
| Angiopoietin-2 (D200) Antibody #50697 | WB | H |
| PDGF Receptor β (28E1) Rabbit mAb #3169 | WB, IP, IHC, IF | H, M, R |
| Phospho-PDGF Receptor β (Tyr751) (17B2) Rabbit mAb #4549 | WB | H, M |
| Endothelial (and other) Cell Markers Most Important Targets: CD31 (PECAM-1), Ki-67 (proliferation marker), α-SMA |
||
| α-Smooth Muscle Actin (α-SMA) (D4K9N) Rabbit mAb #19245 | WB, IP, IHC, IF | H, M, R, Hm, Mk |
| CD31 (PECAM-1) (D8Q9V) Rabbit mAb #77699 | WB, IHC | M |
| CD34 (E2J1K) Rabbit mAb #26233 | WB, IF | M |
| CD34 (ICO115) Mouse mAb #3569 | IHC, F | H |
| Ki-67 (D3B5) Rabbit mAb #9129 | IF, F | H, M, R |
| VE-Cadherin (D87F2) Rabbit mAb #2500 | WB, IP, IF, F | H, B, Pg |
| VE-Cadherin (E6N7A) Rabbit mAb #93467 | WB, IHC | H |
| Downstream Signaling Pathways in Endothelial Cells Most Important Targets: AKT, ERK1/2, Stat3 (total & phosphorylated forms) |
||
| Akt (pan) (C67E7) Rabbit mAb #4691 | WB, IP, IHC, IF, F | H, M, R, Mk, Dm |
| Phospho-Akt (Ser473) (D9E) Rabbit mAb #4060 | WB, IHC, IF, F | H, M, R, Hm, Mk, Dm, Z, B |
| p44/42 MAPK (Erk1/2) (137F5) Rabbit mAb #4695 | WB, IP, IHC, IF, F | H, M, R, Hm, Mk, Mi, Dm, Z, B, Dg, Pg, Ce |
| Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (D13.14.4E) Rabbit mAb #4370 | WB, IP, IHC, IF, F | H, M, R, Hm, Mk, Mi, Dm, Z, B, Dg, Pg, Sc |
| Stat3 (D3Z2G) Rabbit mAb #12640 | WB, IP, IF, F, ChIP | H, M, R, Mk |
| Stat3 (79D7) Rabbit mAb #4904 | WB, IP, ChIP, | H, M, R, Mk |
| Phospho-Stat3 (Tyr705) (D3A7) Rabbit mAb #9145 | WB, IP, IHC, IF, F, ChIP | H, M, R, Mk |
Additional Resources
Read the additional blogs in the Hallmarks of Cancer Series:
- Evading Growth Suppressors
- Nonmutational Epigenetic Reprogramming
- Avoiding Immune Destruction
- Tumor-Promoting Inflammation
- Activating Invasion & Metastasis
- Senescent Cells
- Genome Instability & Mutation
- Resisting Cell Death
- Deregulating Cellular Metabolism
- Unlocking Phenotypic Plasticity
- Sustaining Proliferative Signaling
Select References
- Hanahan D, Weinberg RA (January 2000). "The Hallmarks of Cancer". Cell. 100 (1): 57–70. doi:10.1016/S0092-8674(00)81683-9
- Hanahan D, Weinberg RA (March 2011). "Hallmarks of Cancer: the next generation". Cell. 144 (5):646-74. doi: 10.1016/j.cell.2011.02.013.
- Hanahan D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022;12(1):31-46. doi:10.1158/2159-8290.CD-21-1059
- Liu ZL, et al. Signal Transduct Target Ther. 2023. Angiogenic signaling pathways and anti-angiogenic therapy for cancer.
- Liu X, et al. Biomark Res. 2025 Decoding tumor angiogenesis: pathways, mechanisms, and future directions in anti-cancer strategies.