What is apoptosis?
Apoptosis is a form of programmed cell death (PCD) characterized by cell shrinkage, chromatin condensation, membrane blebbing, and nuclear fragmentation. It is critical for the formation and maintenance of healthy tissue, and nearly all organisms rely on apoptosis during early embryonic development to remove unnecessary cells. It is also a critical part of tissue homeostasis and is used to maintain the balance between cell proliferation and cell death within a population by eliminating old or damaged cells.
When apoptosis is disrupted, however, the consequences can be severe. Excessive apoptosis can contribute to developmental abnormalities and neurodegenerative diseases, while insufficient apoptosis may allow damaged or unwanted cells to survive, promoting cancer progression, autoimmune diseases, and chronic viral infections.


This blog explores the mechanisms, pathways, and proteins involved in apoptosis, as well as key assays and markers for studying its role in health and disease.
Mechanisms of Apoptosis: Intrinsic and Extrinsic Pathways
Apoptosis is a tightly regulated process characterized by distinct morphological changes and the activation of specific caspases and mitochondrial control pathways.
There are two main mechanisms that can trigger apoptosis:


Both the intrinsic and extrinsic pathways activate a complex signaling cascade during which the cell progresses through early, mid- and late-stage apoptosis. The following key events occur in each phase:
- Early Phase: Early apoptosis is marked by either the binding of death ligands and formation of the DISC complex (extrinsic), or by cellular stress activating pro-apoptotic Bcl-2 family proteins (intrinsic). This leads to mitochondrial outer membrane permeabilization (MOMP), activation and translocation of pro-apoptotic proteins (such as Bax and Bak) to the mitochondria, cytochrome c release, and the activation of initiator caspases (caspase-8 for extrinsic, caspase-9 for intrinsic). In parallel, phosphatidylserine translocates to the outer leaflet of the plasma membrane.
- Mid-Phase: Activation of executioner caspases (caspase-3, caspase-6, and caspase-7), which leads to the cleavage of key substrates such as PARP and lamin A/C, cell shrinkage, cytoskeleton collapse, chromatin condensation, and nuclear fragmentation.
- Late Phase: DNA fragmentation, membrane blebbing, and formation of apoptotic bodies, which are then recognized and engulfed by phagocytes.
Because apoptosis shares some overlapping features and pathways with other forms of programmed cell death, such as autophagy and necrosis, multiple markers and assays are typically needed to verify the type and stage of cell death occurring. The following sections describe the mechanisms of apoptosis in more detail to help you identify the most appropriate methods for identifying it in your experimental model.
Detecting Apoptosis: Key Markers & Assays
The TUNEL Assay
During the late stages of apoptosis, DNA is fragmented by an endonuclease, which cleaves chromosomes into nucleosomal units that can be visualized on gels as DNA laddering. DNA fragmentation also serves as the basis for monitoring apoptosis in situ using the TUNEL assay, a robust and widely used method of identifying cell death. The TUNEL assay labels the 3’OH end of fragmented DNA with a modified dUTP conjugated to a fluorophore, which can then be detected using immunofluorescence (IF), immunohistochemistry (IHC), or flow cytometry.
Figure 1 shows fragmented DNA (red) in a developing mouse embryo, where the cells between the digits are undergoing programmed cell death to separate the fingers, a classic example of apoptosis.
 Figure 1. Cells undergoing apoptosis during the separation of the digits in a fixed frozen mouse embryo (E14.5) at low (left) and high magnification (right). The TUNEL Assay Kit (Fluorescence, 594 nm) #48513 assay (red) reveals DNA fragmentation characteristic of apoptosis, while recombinant monoclonal antibody Cleaved Caspase-3 (Asp175) (5A1E) Rabbit mAb #9664 (green) identifies cells executing the intrinsic apoptotic pathway. DAPI #4083 (blue) staining labels all nuclei, showing cellular organization.
Figure 1. Cells undergoing apoptosis during the separation of the digits in a fixed frozen mouse embryo (E14.5) at low (left) and high magnification (right). The TUNEL Assay Kit (Fluorescence, 594 nm) #48513 assay (red) reveals DNA fragmentation characteristic of apoptosis, while recombinant monoclonal antibody Cleaved Caspase-3 (Asp175) (5A1E) Rabbit mAb #9664 (green) identifies cells executing the intrinsic apoptotic pathway. DAPI #4083 (blue) staining labels all nuclei, showing cellular organization.
However, since DNA fragmentation can also occur during necrosis, it is crucial not to rely on TUNEL as an indicator of apoptosis versus other forms of cell death. Morphological analysis can help, as the DNA damage observed in apoptosis is associated with small, round, evenly distributed apoptotic bodies, whereas in necrosis, DNA fragmentation is less organized and associated with cell lysis.

Characterizing Membrane Changes
Another way to detect apoptosis is to visualize changes to the cell membrane. Notably, the cell membrane remains intact during the early and mid-stages of apoptosis, which prevents damage and inflammation to surrounding tissues. As a result, intracellular cytokines and digestive enzymes are not released into the extracellular space—a characteristic that can aid in the identification of apoptosis compared to other forms of cell death.
A hallmark of apoptosis is when phosphatidylserine lipids within the cell membrane, which normally face the cytosol, flip to the cell surface and become exposed to the extracellular space. Their presence acts as an “eat me” signal for phagocytes to remove the dead cell. Due to its ability to bind to phosphatidylserine lipids on the cell membrane, Annexin V is often used as a marker of early apoptosis. However, since phosphatidylserine is also exposed in lysed cells during necrosis, or when the membrane becomes permeable in late apoptosis, it is important to perform Annexin V staining in combination with vital dyes such as propidium iodide (PI), as shown in Figure 2. PI is a fluorescent DNA dye that only stains when the membrane is compromised during necrosis or the late stages of apoptosis.
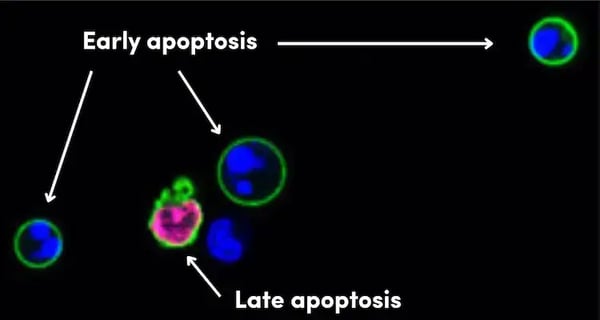
Figure 2. IF analysis of live Jurkat cells treated with camptothecin, an apoptosis inducer, using Annexin V-FITC Early Apoptosis Detection Kit #6592 (green), which labels cells with externalized phosphatidylserine—a hallmark of early apoptosis. Propidium iodide (red) shows a cell with a permeabilized membrane, which occurs during necrosis or late-stage apoptosis. DRAQ5® #4084 (blue) marks all nuclear DNA.

The Bcl-2 Family Proteins in Apoptosis
The intrinsic (mitochondrial) apoptotic pathway involves many conserved signaling proteins and depends upon the integrity of the mitochondria. The process is tightly regulated by a balance between the activity of proteins in the Bcl-2 family, which consists of pro-apoptotic (Bax, Bak, Bad, Bid, Puma, Bim, and Noxa) and anti-apoptotic factors (Bcl-2, Bcl-xL, Bcl-w, A1/Bfl-1, and Mcl-1).
The pro-apoptotic Bcl-2 family members are divided into two groups: The multidomain executioners, such as Bax and Bak, which contain several Bcl-2 homology (BH) domains (BH1, BH2, and BH3), and the BH3-only proteins, including Bad, Bid, Puma, Bim, and Noxa, which contain only the BH3 domain. (Note: Bid can also become activated in the extrinsic pathway to amplify the apoptotic signal.)
The interaction between these two groups and the anti-apoptotic proteins is mediated by their BH domains—the BH3-only proteins can bind to anti-apoptotic Bcl-2 family members, neutralizing their function and allowing activation of Bax and Bak, which leads to mitochondrial outer membrane permeabilization and apoptosis.
This interaction is therapeutically targeted in cancer by BH3 mimetics—small molecules that mimic the action of BH3-only proteins. For example, the Bcl-2 inhibitor Venetoclax binds to anti-apoptotic Bcl-2 family proteins, displacing pro-apoptotic proteins and promoting cell death in cancer cells.

During times of cellular stress or in response to post-translational modifications, BH3-only proteins become activated and translocate to the mitochondria, where they interact with and activate the multidomain executioner proteins Bax and Bak. The co-localization of pro-apoptotic proteins at the mitochondria, such as Bim and Bak in Figure 3, is a hallmark of apoptosis initiation, as it marks the critical step where the cell commits to self-destruction.
 Figure 3. Top: IF analysis using recombinant monoclonal antibody Bim (C34C5) Rabbit mAb #2933 (green) showing colocalization of BIM with mitochondria. Bottom: IF analysis using recombinant monoclonal antibody Bak (D4E4) Rabbit mAb #12105 (green) showing colocalization of BAK with mitochondria. In both figures, mitochondria have been labeled with MitoTracker Red CMXRos #9082 (red) and DRAQ5® #4084 (blue) marks all nuclear DNA.
Figure 3. Top: IF analysis using recombinant monoclonal antibody Bim (C34C5) Rabbit mAb #2933 (green) showing colocalization of BIM with mitochondria. Bottom: IF analysis using recombinant monoclonal antibody Bak (D4E4) Rabbit mAb #12105 (green) showing colocalization of BAK with mitochondria. In both figures, mitochondria have been labeled with MitoTracker Red CMXRos #9082 (red) and DRAQ5® #4084 (blue) marks all nuclear DNA.
After Bax and Bak become activated, they induce changes in the mitochondrial outer membrane permeability (MOMP), leading to the release of cytochrome c, a hallmark of the intrinsic (mitochondrial) apoptotic pathway. Free cytochrome c in the cytoplasm then interacts with Apaf-1 to form a complex called the apoptosome, which activates caspase-9 and triggers a cascade of apoptotic events that dismantle the cell.
Another way of identifying apoptosis is to measure mitochondrial membrane potential to identify the occurrence of MOMP. This type of assay uses a mitochondrial membrane-permeable fluorescent dye, such as TMRE (tetramethylrhodamine ethyl ester perchlorate), which accumulates in healthy, polarized mitochondria. When the mitochondrial membrane potential is lost—an early event in apoptosis—TMRE fluorescence decreases, indicating mitochondrial dysfunction. The Mitochondrial Membrane Potential Assay Kit (II) #13296 from CST includes TMRE, along with a control compound, CCCP, which uncouples mitochondrial oxidative phosphorylation and leads to depolarization of the mitochondrial membrane.
It is important to note, however, that the loss of mitochondrial membrane potential, and associated loss of TMRE staining, can also occur in necrotic cell death, so additional assays are needed to distinguish between the two.
The Role of Caspases in Apoptosis
Both the intrinsic and extrinsic apoptotic pathways are ultimately dependent on the activity of caspases, a family of cysteine proteases that play essential roles in programmed cell death. Caspases can be broadly categorized based on their function:
- Initiator caspases, including caspase-2, caspase-8, caspase-9, caspase-10, and caspase-12, which are activated in response to pro-apoptotic signals. When activated, initiator caspases cleave and activate downstream executioner caspases.
- Executioner caspases, including caspase-3, caspase-6, and caspase-7, are responsible for dismantling the cell by cleaving key structural and regulatory proteins during apoptosis.
In addition to their roles in apoptosis, certain caspase family members—such as caspase-1, caspase-4, caspase-5, and caspase-11—are primarily involved in inflammation and pyroptosis, a distinct form of programmed cell death.

In the extrinsic pathway, caspases are activated when extracellular ligands bind to cell surface death receptors in the TNFR family (Fas, TNF-R1,TNF-R2, DR3, DR4, and DR5), and their associated ligands, which include TNF-α, FasL, TRAIL, and TWEAK. Ligand binding induces receptor activation, which leads to the formation of a Death-Inducing Signaling Complex (DISC), which recruits and cleaves the initiator caspase-8 to activate it. Identifying the presence of caspase-8 can indicate if apoptosis can occur in a cellular population, as shown in Figure 4.
 Figure 4. Immunohistochemical (IHC) analysis of paraffin-embedded human endometrioid adenocarcinoma using Caspase-8 (F5K9P) Rabbit mAb #8873. This antibody stains both full-length and cleaved caspase-8, indicating that caspase-8-dependent apoptosis can occur in these cells.
Figure 4. Immunohistochemical (IHC) analysis of paraffin-embedded human endometrioid adenocarcinoma using Caspase-8 (F5K9P) Rabbit mAb #8873. This antibody stains both full-length and cleaved caspase-8, indicating that caspase-8-dependent apoptosis can occur in these cells.
Cleaved caspase-8 then cleaves and activates the executioner caspases, such as caspase-3 and caspase-7, which orchestrate the cleavage of cellular substrates and the morphological changes characteristic of apoptosis. Cleavage-specific antibodies targeting active forms of caspases or their substrates are important tools for identifying apoptotic cell death. For example, detection of cleaved caspase-8 is a key marker of apoptotic cell death via the extrinsic pathway, as shown in Figure 5.
 Figure 5. IF analysis of Raw 264.7 cells, untreated (left) or treated with TNF-α and Cycloheximide, using Cleaved Caspase-8 (Asp387) (D5B2) XP® Rabbit mAb #8592 (green), which labels cells undergoing caspase-8 activation—a hallmark of extrinsic apoptosis. Blue pseudocolor = DRAQ5® #4084 (fluorescent DNA dye).
Figure 5. IF analysis of Raw 264.7 cells, untreated (left) or treated with TNF-α and Cycloheximide, using Cleaved Caspase-8 (Asp387) (D5B2) XP® Rabbit mAb #8592 (green), which labels cells undergoing caspase-8 activation—a hallmark of extrinsic apoptosis. Blue pseudocolor = DRAQ5® #4084 (fluorescent DNA dye).
In the intrinsic pathway, caspase-9 is the primary initiator caspase. Unlike the extrinsic pathway, which relies on death receptors, the intrinsic pathway is dependent on mitochondrial activation of the Bcl-2 family members, as described above. Once activated, caspase-9 triggers a cascade that cleaves and activates the executioner caspases, leading to the demolition of the cell.
Since they are conserved between both pathways, Cleaved PARP, cleaved lamin A/C, and cleaved caspase-3, shown in Figure 6, are well-established markers of caspase activity and apoptosis activation.
Figure 6. IF analysis of HT-29 cells, untreated (left) or Staurosporine #9953 treated (right) and labeled with recombinant monoclonal antibody Cleaved Caspase-3 (Asp175) (5A1E) Rabbit mAb #9664 (green). Actin filaments have been labeled with Alexa Fluor® 555 phalloidin #8953 (red). Blue pseudocolor = DRAQ5® #4084 (fluorescent DNA dye).
Caspase Inhibition by IAP Family Proteins
Proteins in the Inhibitors of Apoptosis (IAP) family, which include XIAP, c-IAP1, c-IAP2, NAIP, Livin, and Survivin, can block the activity of different caspases to inhibit apoptosis.
For example, XIAP binds to caspase-3, caspase-7, and caspase-9 to inhibit their activity and prevent the cleavage of key apoptotic proteins. The expression of IAP family members is an indicator of enhanced cell survival.
Interaction between the IAP family and caspases occurs via a conserved baculoviral IAP repeat (BIR) domain. Notably, some IAP family members, such as c-IAP1, c-IAP2, and XIAP, also contain a RING domain that confers ubiquitin E3 ligase activity. Through their RING domain, these proteins can ubiquitinate and promote the degradation of several important signaling proteins, including those within the canonical TNF death receptor pathway, thereby regulating both cell death and inflammation.
The extrinsic cell death receptor pathway can also lead to cell survival through TNFR2-mediated signaling to NF-κB, which induces expression of pro-survival genes, Bcl-2 and FLIP.
Additional Resources
To learn more about the mechanisms, morphology, and key proteins involved in many types of cell death, download the guide below:

Read the additional blog posts in the Mechanisms of Cell Death series:
- Mechanisms of Cell Death: Ferroptosis
- Mechanisms of Cell Death: Pyroptosis
- Mechanisms of Cell Death: Necrosis & Necroptosis
Select References
- Dickens LS, Powley IR, Hughes MA, MacFarlane M. The 'complexities' of life and death: death receptor signalling platforms. Exp Cell Res. 2012;318(11):1269-1277. doi:10.1016/j.yexcr.2012.04.005
- Favaloro B, Allocati N, Graziano V, Di Ilio C, De Laurenzi V. Role of apoptosis in disease. Aging (Albany NY). 2012;4(5):330-349. doi:10.18632/aging.100459
- Diepstraten ST, Anderson MA, Czabotar PE, Lessene G, Strasser A, Kelly GL. The manipulation of apoptosis for cancer therapy using BH3-mimetic drugs. Nat Rev Cancer. 2022;22(1):45-64. doi:10.1038/s41568-021-00407-4
- McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease [published correction appears in Cold Spring Harb Perspect Biol. 2015 Apr;7(4). pii: a026716. doi: 10.1101/cshperspect.a026716]. Cold Spring Harb Perspect Biol. 2013;5(4):a008656. Published 2013 Apr 1. doi:10.1101/cshperspect.a008656
- Shamas-Din A, Kale J, Leber B, Andrews DW. Mechanisms of action of Bcl-2 family proteins. Cold Spring Harb Perspect Biol. 2013;5(4):a008714. Published 2013 Apr 1. doi:10.1101/cshperspect.a008714
Updated June 2025. Originally published March 2021. 20-CEP-9437 and 25-HMC-18550