Imagine driving down the highway with no brakes. That is essentially what happens when tumor cells lose the molecular “braking systems” that normally restrain cell proliferation to maintain tissue homeostasis.
In the cancer hallmark Evading Growth Suppressors, cancer cells disable multiple tumor suppressor pathways that control cell cycle progression, cell growth, regulation of DNA damage checkpoints and repair mechanisms, and the induction of apoptotic cell death. The most well-studied of these—including Rb, p53, PTEN, and CDKN2—are covered in this blog post.
< Jump to the product list at the end of this blog >
Hitting the Brakes: How Tumor Suppressors Work
|
Under normal conditions, tumor suppressors act as “brakes” on cell growth—key checkpoints that slow or stop proliferation when cells encounter stress, replication errors, or oncogenic signaling. These suppressors regulate checkpoints throughout the cell cycle, helping to maintain homeostasis by enforcing cell cycle arrest, coordinating DNA damage repair, and triggering programmed cell death (apoptosis) when damage is irreparable. |
 Evading Growth Suppressors is one of the original six cancer hallmarks, first described in 2000.
Evading Growth Suppressors is one of the original six cancer hallmarks, first described in 2000.Tumor progression frequently involves the inactivation of these safeguards, often requiring a “double hit” of inherited germline mutation and somatic alterations that disable growth‑restricting pathways. Among the roughly 70–80 tumor suppressors implicated in cell proliferation and survival, Rb, p53, PTEN, and CDKN2A (encoding p16 INK4A and p14 ARF) are among the best characterized and most frequently altered across diverse cancer types.
Relevant pathways that intersect with these tumor suppressors include:
 |
PI3K/Akt Signaling PathwayIntegrates growth factor cues to promote survival, metabolism, and proliferation. PTEN constrains this pathway by antagonizing PIP3 accumulation and downstream Akt activation. Get the PI3K/Akt Signaling pathway diagram |
 |
The G1/S CheckpointControlled in part by Rb, CDKs, and CDK inhibitors such as p16, this checkpoint determines whether cells commit to DNA replication in response to mitogenic and anti‑growth signals. Get the G1/S Checkpoint pathway diagram |
 |
DNA Damage and p53 Signalingp53 and its downstream targets, including p21 and GADD45, help coordinate DNA repair with cell cycle arrest before mitosis, limiting the propagation of genomic lesions. Get the DNA Damage and p53 Signaling pathway diagram |
 |
Wnt/β‑Catenin Signaling PathwayAPC and related regulators restrain β‑catenin–driven transcriptional programs that otherwise enhance proliferative and stem‑like behavior in many epithelial tumors. Get the Wnt/β-Catenin Signaling pathway diagram |
 |
mTOR Signaling PathwayA central node for nutrient and growth factor sensing, PI3K/Akt activation and PTEN loss converge on mTOR to drive growth and metabolic reprogramming. Get the mTOR Signaling pathway diagram |
Rb: The Gatekeeper of the G1/S Checkpoint
The retinoblastoma (Rb) protein was the first tumor suppressor identified, following the discovery that germline mutations in Rb result in retinoblastoma, a rare pediatric eye cancer. At the molecular level, Rb binds to the E2F family of transcription factors to block the recruitment of transcriptional co-activators and repress the expression of genes required for G1-to-S phase transition.
In normal cells, mitogenic cues activate cyclin‑dependent kinases (CDKs), which hyperphosphorylate Rb, leading to its dissociation from E2F and allowing E2F-mediated gene regulation and cell cycle progression. In many cancers, this brake is effectively removed either through mutations in Rb itself or through mutations in other genes in the pathway. For example, CDK overexpression or silencing of the CDK inhibitor p16 INK4A can lead to Rb inactivation, resulting in constitutive E2F activity and uncontrolled proliferation.

Immunohistochemical (IHC) analysis of paraffin-embedded human colon carcinoma using Phospho-Rb (Ser807/811) (D20B12) Rabbit Monoclonal Antibody #8516.
The p53 Tumor Suppressor
The p53 tumor suppressor functions as a stress‑responsive transcription factor that integrates signals from DNA damage, oncogene activation, hypoxia, and other cellular stresses to determine cell fate. Once activated, p53 induces the transcription of different sets of target genes depending on the type and severity of the stress—for example, the transcription of p21 Waf1/Cip1 and GADD45 regulate the cell cycle to give cells time for DNA repair, whereas pro-apoptotic factors including Bax and Puma are transcribed to eliminate cells if the damage is more severe or persistent.
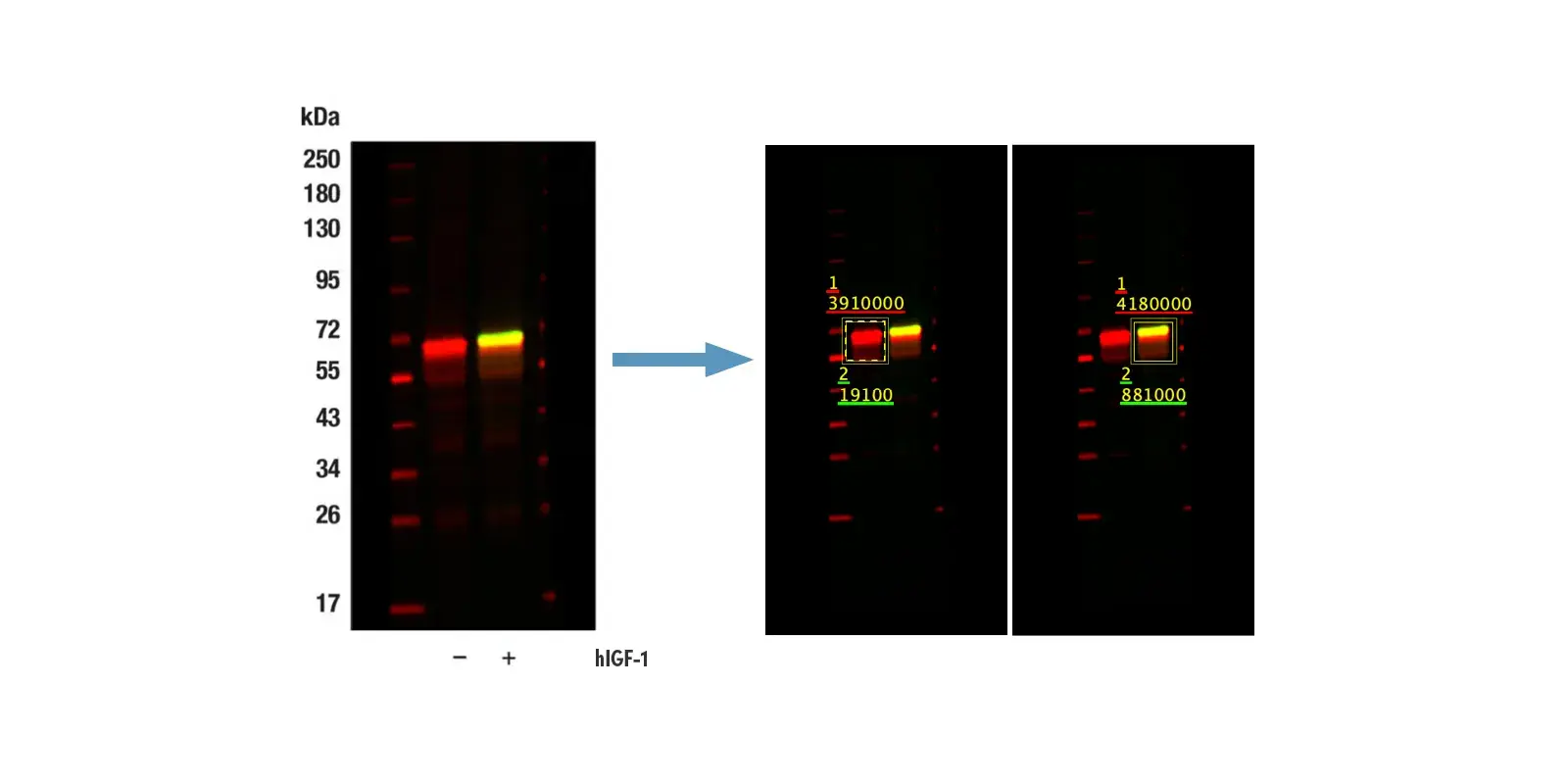
In about half of all human cancers, p53 is inactivated by mutation or deletion, compromising DNA damage responses, increasing the ability of tumors to resist cell death, and leading to genomic instability, which accelerates the acquisition of additional oncogenic alterations. p53 can also be regulated by the ubiquitin E3 ligase MDM2, which targets p53 for degradation—therefore, increased MDM2 expression or activity can result in the silencing of p53 without mutation to the gene itself. Additionally, post-translational modifications, including phosphorylation and acetylation, regulate p53 stability, localization, and DNA binding. For example, the phosphorylation of p53 at Ser15 following DNA damage decreases its association with MDM2, leading to p53 accumulation. However, in many tumors, although p53 may still accumulate through this mechanism, mutations in its DNA‑binding domain render it unable to activate target genes, allowing damaged cells to continue proliferating. 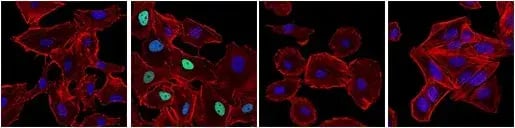
IF analysis of A549 cells (p53-positive), untreated (first image), treated with Doxorubicin #5927 (0.5 µM, 24 hr; second image), or treated with Doxorubicin and post-processed with λ-phosphatase (third image). Saos-2 cells (p53-negative) were treated with Doxorubicin to verify specificity for p53 (fourth image). Cells were labeled with Phospho-p53 (Ser15) (E9Y4U) Rabbit Monoclonal Antibody #82530 (green) and DyLight® 554 Phalloidin #13054 (red) before mounting in ProLong® Gold Antifade Reagent with DAPI #8961 (blue).
The Multi-Faceted Role of CDKN2A
CDKN2A, another important gene in tumor suppression, is inactivated in about 40% of human cancers. This locus encodes two tumor suppressors, p16 INK4A and p14 ARF, which are generated via alternative transcription factors and control cell cycle progression through complementary mechanisms.
p16 inhibits the activity of cyclin-dependent kinases, preventing their association with cyclin D and thereby blocking Rb phosphorylation and G1 progression. In parallel, p14 ARF binds to and sequesters MDM2, stabilizing p53 and promoting the induction of downstream targets such as p21 and other pro-apoptotic genes, leading to cell cycle arrest and, when damage is severe, apoptosis.
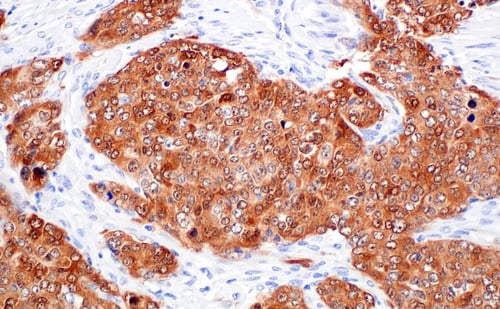
IHC analysis of paraffin-embedded human ovarian serous papillary carcinoma using p16 INK4A (F9T1L) Rabbit Monoclonal Antibody #88667.
Because CDKN2A converges on both the Rb and p53 pathways, CDKN2A mutations or loss can simultaneously compromise cell cycle checkpoints and apoptotic responses, providing a powerful growth advantage to tumor cells. This dual impact makes CDKN2A a critical node in the network of growth suppressors.
PTEN Mutation & Loss
The phosphoinositide 3-kinase (PI3K)/Akt signaling pathway is a key driver of tumor growth, promoting survival, cell growth, and metabolic reprogramming, in part through downstream activation of mTOR. PTEN encodes a lipid phosphatase that dephosphorylates phosphatidylinositol (3,4,5)-trisphosphate (PIP3) to generate phosphatidylinositol (4,5)-bisphosphate (PIP2), thereby suppressing PI3K and dampening Akt activation.
When PTEN is mutated, deleted, or otherwise inactivated, as is the case in many cancers, it results in sustained Akt and mTOR pathway activation, which supports proliferation, survival, and metabolic reprogramming in cancer cells. Consequently, PTEN‑deficient tumors can display aggressive clinical behavior and may show resistance to therapies that rely on intact apoptotic responses or that fail to target the PI3K/Akt/mTOR axis.
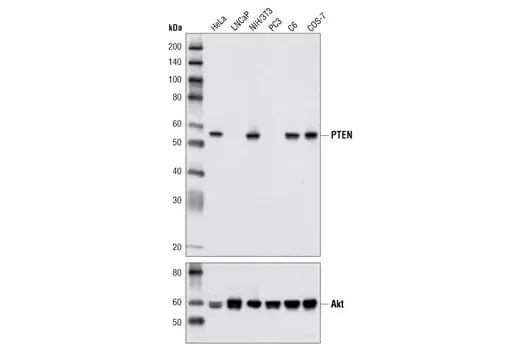
Western blot analysis of extracts from various cell lines using PTEN (D4.3) Rabbit Monoclonal Antibody #9188 (upper) and Akt (pan) (C67E7) Rabbit Monoclonal Antibody #4691 (lower).
Other Tumor Suppressors
There are many other well-described tumor suppressors involved in tumor progression, frequently associated with specific tumor types and tissues. These include:
- BRCA1/2: Mutations in Breast cancer type 1 susceptibility protein 1 and 2 (BRCA1 and BRCA2) increase susceptibility to breast and ovarian cancer; these proteins are involved in high-fidelity DNA repair, so mutations result in increased genomic instability.
- APC: Adenomatous polyposis coli (APC) is mutated in most familial and sporadic colorectal cancers and is important for controlling the degradation of β-Catenin, a key downstream effector in the Wnt signaling pathway, so an absence of APC leads to increased proliferative signaling.

IHC analysis of paraffin-embedded human prostate adenocarcinoma using beta-Catenin (D10A8) Rabbit Monoclonal Antibody #8480 performed on the Leica BOND Rx. In some cancers, mutations in APC prevent the degradation of ß-Catenin, leading to increased proliferative signaling.
- KEAP1: Mutations in Kelch-like ECH-Associated protein 1 (KEAP1) occur in non-small cell lung cancer (NSCLC) and disrupt the regulation of the transcription factor NRF2, leading to altered oxidative stress responses and metabolic adaptations that support tumor growth.
- LKB1: Liver kinase B1 (LKB1) is a serine/threonine kinase that regulates metabolic signaling, including AMPK. Its loss contributes to the metabolic adaptations that drive cancer progression.
- NF1: Mutations in Neurofibromin 1 (NF1), which negatively regulates Ras, can result in the over-activation of the Ras/MAPK signaling pathway and are a feature of neurofibromas and other tumors.
Therapeutic Opportunities for When the Brakes Fail
A major challenge in oncology is that tumor suppressors are often lost or inactivated, making them difficult to restore directly with conventional small molecules. Loss of these safeguards can also reduce sensitivity to DNA‑damaging chemotherapies or radiation, as seen with tumors harboring dysfunctional p53 genes that fail to undergo apoptosis after treatment.
To address this, many strategies focus on exploiting synthetic lethality or targeting downstream vulnerabilities created by tumor suppressor loss. A classic example is the use of PARP inhibitors in tumors with BRCA1/2 defects, where inhibition of PARP‑mediated DNA repair selectively kills BRCA‑deficient cells while sparing those with intact homologous recombination.
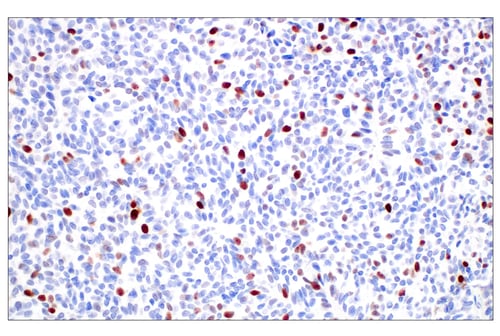
IHC analysis of paraffin-embedded human papillary carcinoma of the breast using BRCA1 (E5S9G) Rabbit Monoclonal Antibody #50799.
As our understanding of tumor suppressor networks deepens, new therapeutic avenues are emerging, including restoring p53 activity, targeting hyperactive PI3K/Akt/mTOR signaling in PTEN‑deficient tumors, and modulating the metabolic and redox pathways altered by KEAP1 or LKB1 mutations. For researchers studying normal biology in the context of disease, dissecting how these pathways intersect at key nodes like Rb, p53, CDKN2A, and PTEN remains essential for uncovering new biomarkers and rational combination therapies.
| p53 Antibody Sampler Kit #37909 |
| Phospho-p53 Antibody Sampler Kit #65390 |
| Rb Antibody Sampler Kit #9969 |
Additional Resources
Read the additional blogs in the Hallmarks of Cancer Series:
- Nonmutational Epigenetic Reprogramming
- Avoiding Immune Destruction
- Tumor-Promoting Inflammation
- Activating Invasion & Metastasis
- Inducing or Accessing Vasculature (Angiogenesis)
- Senescent Cells
- Genome Instability & Mutation
- Resisting Cell Death
- Deregulating Cellular Metabolism
- Unlocking Phenotypic Plasticity
- Sustaining Proliferative Signaling
Select References
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57-70. doi:10.1016/s0092-8674(00)81683-9
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-674. doi:10.1016/j.cell.2011.02.013
- Hanahan D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022;12(1):31-46. doi:10.1158/2159-8290.CD-21-1059
- Amin ARMR, Karpowicz PA, Carey TE, et al. Evasion of anti-growth signaling: A key step in tumorigenesis and potential target for treatment and prophylaxis by natural compounds. Semin Cancer Biol. 2015;35 Suppl:S55-S77. doi:10.1016/j.semcancer.2015.02.005
- Scheel CH, Schäfer R. Editorial: Hallmark of cancer: Evasion of growth suppressors. Front Oncol. 2023;13:1170115. Published 2023 Mar 13. doi:10.3389/fonc.2023.1170115
- Kuo KK, Hsiao PJ, Chang WT, et al. Therapeutic Strategies Targeting Tumor Suppressor Genes in Pancreatic Cancer. Cancers (Basel). 2021;13(15):3920. Published 2021 Aug 3. doi:10.3390/cancers13153920
- Lea M. TUMOR SUPPRESSOR GENES. Molecular Oncology. 2012.