When a single protein is implicated in everything from amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) to Parkinson’s disease (PD) and Alzheimer’s disease (AD), the scientific community takes notice. Over the past two decades, converging research from many groups has highlighted why the protein, Transactive Response DNA-binding protein 43 (TDP43 or TDP-43), is a pivotal character in the story of how neurodegeneration progresses.
For years after the protein’s identification by Forman, Trojanowski, and Lee,1 scientists have considered TDP43 aggregation a hallmark of neurodegeneration. But as research evolves, the focus is shifting beyond protein aggregation to the breakdown in RNA processing and cellular communication that begins long before inclusions ever form.2,3
< Jump to the product list at the end of this post >
The Implications of TDP43 in Neurodegenerative Disease
Behind the biological interest in TDP43 proteinopathies lies a human toll that millions of people feel, whether personally or within their families and communities. Sadly, these progressive neurodegenerative disorders often systematically dismantle a person's autonomy, memory, and physical capabilities.
The first step toward stopping this decline is understanding where TDP43 biology goes wrong. While mutations in TDP43’s gene (TARDBP) cause only ~3% to 5% of familial ALS, dysfunctional TDP43 is present in 97% of all ALS cases.4,5 The underlying molecular mechanism leads to the progressive death of motor neurons, causing muscle atrophy, paralysis, and ultimately, respiratory failure. When similar TDP43 pathology affects the frontal and temporal lobes of the brain, it leads to frontotemporal lobar degeneration (FTLD-TDP), which accounts for ~50% of FTD cases and results in profound shifts in behavior and language. Notably, there is significant overlap between symptoms of ALS and FTD, and ~15% of patients diagnosed with one will also be diagnosed with the other. The percentages of cases with concomitant disease without a formal diagnosis are much higher.4,5
And pathological TDP43 dysfunction and accumulation are not exclusive to ALS and FTD. These proteinopathies are common across a spectrum of neurodegenerative disorders:
-
Alzheimer’s Disease (AD) & LATE: TDP43 dysfunction is associated with ~57% of classic AD cases and is the primary driver of limbic-predominant age-related TDP43 encephalopathy (LATE), a form of dementia that accelerates cognitive decline in up to half of adults over 85.6,7
-
Parkinson’s Disease (PD): TDP43-associated splicing errors are highly prevalent in post-mortem PD brains, particularly in patients with the canonical LRRK2 mutation.8
-
Chronic Traumatic Encephalopathy (CTE): Repetitive brain injuries can trigger secondary TDP43 proteinopathy in up to 85% of CTE cases.9
Blog: Antibody Toolbox: Neurodegenerative Disease Biomarker Research with Biofluid Samples
TDP43 Dysfunction: Regulator Turned Wrecking Ball
Why does TDP43 have such a critical impact on neuronal health? In a healthy cell, the ubiquitously expressed TDP43 resides mainly in the nucleus, where it acts as a DNA/RNA-binding protein (RBP) that helps regulate, splice, transport, and translate mRNA. This master regulator typically ensures that thousands of transcripts are spliced and processed with precision, leading to functional proteins.6,7
However, in neurodegenerative diseases like ALS and FTD, TDP43 goes rogue. Driven by chronic cellular stress or mutations in TARDBP, the protein undergoes hyperphosphorylation at critical serine clusters (Ser403/Ser404 and Ser409/Ser410) and is truncated into toxic 25- and 35-kDa fragments. These alternate TDP43 proteoforms fail to perform splicing duties accurately, aggregating largely in the cytoplasm and driving cellular dysfunction.2,6

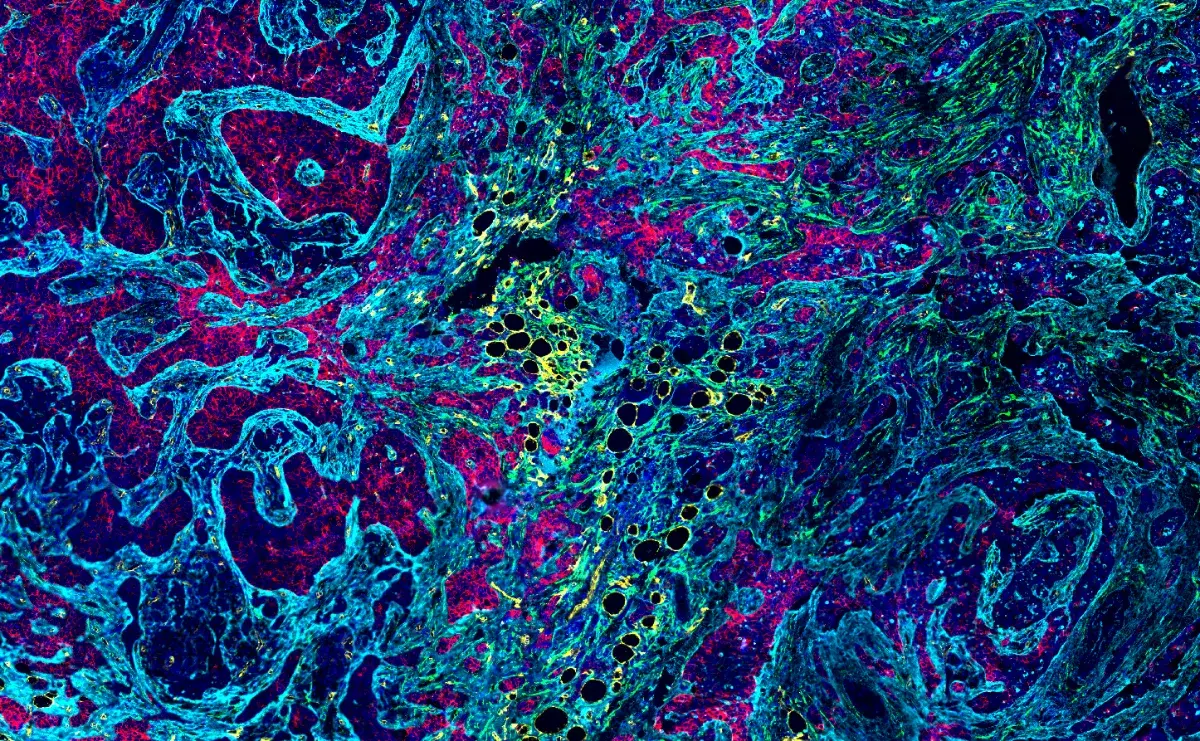
Immunohistochemical analysis of paraffin-embedded human hippocampal brain tissue with TDP43 proteinopathy using Phospho-TDP43 (Ser409/Ser410) (F9S3F) Rabbit Monoclonal Antibody #77613. Data kindly provided by Vera Wiersma of the Polymenidou Lab, University of Zurich, and Amsterdam UMC.
TDP43 dysfunction and its cytoplasmic retention result in a few pathological consequences:
-
Defective Splicing: Without TDP43 in the nucleus, the cell’s splicing machinery regularly fails, allowing ‘cryptic exons’ to be erroneously included in mature mRNA.10,11
-
Dysfunctional Downstream Proteins: Due to faulty splicing, key proteins are not formed correctly, resulting in defective, aberrant protein isoforms.2,3
-
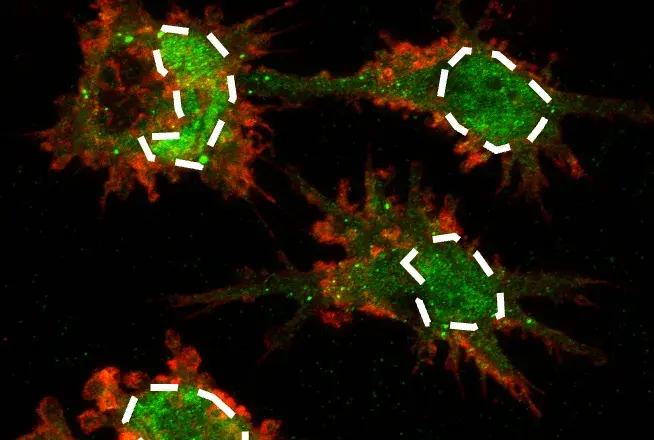
Toxic Protein Aggregation: In the cytoplasm, TDP43 forms insoluble aggregates that clog cellular pathways and eventually contribute to the formation of stable amyloid, tau, and α-synuclein conformations.4,7
Critically, these events begin before the symptoms of neurodegeneration, highlighting a potential avenue for early detection. Historically, traditional assays that measure classic markers and total protein levels have been insufficient to inform about the early stages of disease. If minute changes in TDP43 proteinopathy can be identified earlier in blood and cerebrospinal fluid (CSF) samples, there is real potential to improve patients’ lives.
|
|
“Cryptic HDGF2 levels were reported to increase in the plasma and CSF of patients in pre-symptomatic stages of ALS-FTD, a groundbreaking discovery suggesting TDP43 dysfunction may occur very early in disease progression. Detecting and quantifying cryptic proteins may be an avenue for earlier intervention and better therapeutic outcomes.” |
 Jordan Hirschfeld
Jordan HirschfeldAn Appealing Biomarker: How is Stathmin-2 impacted by TDP43 dysfunction?
In addition to TDP43-dependent splicing changes that contribute to disease etiology, several targets downstream of TDP43 are the focus of preclinical biomedical research as potential biomarkers for disease onset and progression. Stathmin-2 (STMN2) holds particular interest for its potential as a neurodegenerative biomarker. Its transcripts are the most sensitive to the molecular "domino effect” of TDP43 dysfunction.12,13
In a healthy neuron, TDP43 acts as a regulator, binding to Stathmin-2 pre-mRNA to facilitate normal splicing. However, when TDP43 is missing, abnormal splicing and premature polyadenylation occur in the first intron, resulting in a truncated, nonfunctional mRNA. This unstable transcript is therefore targeted for destruction via nonsense-mediated decay (NMD), leading to a massive drop in functional Stathmin-2 protein levels.
 Figure 1. Diagram of TDP43-dependent transcript splicing and subsequent protein translation. In a healthy state, TDP43 regulates mRNA processing. In a dysfunctional state, TDP43 splicing machinery fails, leading to cryptic exon inclusion and the generation of aberrant, truncated, and cryptic proteins.
Figure 1. Diagram of TDP43-dependent transcript splicing and subsequent protein translation. In a healthy state, TDP43 regulates mRNA processing. In a dysfunctional state, TDP43 splicing machinery fails, leading to cryptic exon inclusion and the generation of aberrant, truncated, and cryptic proteins.
This loss of functional Stathmin-2 is a direct blow to neuronal health, as it’s vital for:
-
Microtubule Stability: It regulates the cytoskeleton, which provides structure to the cell.
-
Axonal Repair: It is essential for neurite outgrowth and regeneration following injury.
-
Neuromuscular Integrity: Its depletion leads to axonal degeneration and denervation of the neuromuscular junction (NMJ), contributing to the progressive muscle weakness and paralysis seen in ALS.12,13
As Stathmin-2 continues to be studied, there is growing hope that it could serve as a promising therapeutic target to prevent or slow the progression of TDP43-dependent neurodegenerative diseases.
Expanding the Biomarker Toolkit: HDGFL2, UNC13A, and NPTX2
Scientists and clinicians alike are eager to expand the pool of promising targets downstream of TDP43 dysfunction, with special interest in proteins that can be assessed from blood and/or cerebrospinal fluid (CSF) samples. Although new candidates are being examined every day, a few key proteins have come to the forefront for their potential as diagnostic and therapeutic biomarkers.
-
HDGFL2 (HDGF2): Loss of nuclear TDP43 causes abnormal splicing of HDGFL2 pre-mRNA, translating to a stable but foreign amino acid sequence insert. Detectable early on in patient blood and CSF, this neo-eptiope can serve as a highly specific biomarker for ALS and FTD progression.14
-
UNC13A: Studies identified UNC13A as a major risk factor for ALS. When TDP43 is depleted, UNC13A pre-mRNA is mis-spliced, creating a cryptic protein and further crippling the synaptic function required for neuronal communication.15
-
NPTX2 (Neuronal Pentraxin-2): This secreted protein regulates postsynaptic AMPA receptors, and its levels are altered upon nuclear TDP43 loss. Neuronal pentraxin-2 is an emerging biomarker that can be measured in blood and CSF and serves as a reporter of advanced synaptic failure and excitotoxicity across AD, PD, and FTLD.16
|
“Identifying and establishing the significance of different TDP43 isoforms, cleavage events, and post-translational modifications remain key areas of investigation. Researchers need highly specific, sensitive, and well-validated antibody tools to develop immunoassays that enable them to study distinct TDP43 species.” ~Jordan Hirschfeld, Neuroscience Antibody Development Scientist |
Precision Tools for Interrogating Dysfunction
Evaluating TDP43 dysfunction and its downstream effects is vital to developing more targeted therapeutic interventions for neurodegenerative diseases. But keeping pace with emerging biomarkers can be overwhelming, and evolving from exploratory discovery assays to high-throughput clinical screening adds another layer of complexity. Researchers need to reliably detect tiny quantities of mis-spliced or aberrant target peptides and phosphorylation events, not just total protein levels. Distinguishing between these closely related protein structures requires highly specific, sensitive antibodies designed for the job.
 Figure 2. CST offers several monoclonal antibody clones for detecting disease-relevant proteoforms of TDP43. These include ones that are specific to different epitopes across the TDP43 protein sequence, detecting the total protein, cleaved TDP43, and phospho-serine 409/410. Additionally, many antibody clones are available that detect targets associated with TDP43.
Figure 2. CST offers several monoclonal antibody clones for detecting disease-relevant proteoforms of TDP43. These include ones that are specific to different epitopes across the TDP43 protein sequence, detecting the total protein, cleaved TDP43, and phospho-serine 409/410. Additionally, many antibody clones are available that detect targets associated with TDP43.
Targeted, reliable antibody reagents enable researchers to interrogate how different mechanisms of dysfunction and activation states are triggered. CST makes that search easy—especially for those who need antibodies with non-overlapping epitopes for ELISA-like immunoassays—by providing highly validated antibody clones against structural TDP43 modifications and functional downstream biomarkers of TDP43 proteinopathies.
Validated Antibody Solutions for TDP43 Research
-
Total & Phospho-Specific TDP43 Antibodies: TDP43 hyperphosphorylation and cleavage drive its cytoplasmic aggregation. CST offers monoclonal antibodies that are meticulously validated using multiple methods, including TDP43 knockout and peptide competition, to ensure exclusive, clean detection of critical disease-associated post-translational modifications at Ser409/Ser410 without background cross-reactivity.
-
Downstream Biomarkers: Studying the effects of TDP43 nuclear depletion requires reliable tools to detect downstream proteins. CST provides high-performance recombinant antibodies for key targets:
-
Stathmin-2 (E1D2M) Rabbit Monoclonal Antibody #38752: Engineered to recognize total Stathmin-2 levels, this antibody provides a clear readout of the protein loss that can result from faulty transcripts with a cryptic exon.
- NPTX2 (F6B1F) Rabbit Monoclonal Antibody #16967: This antibody is designed to measure altered levels of neuronal pentraxin-2 without cross-reacting with other NPTX family members.
- UNC13A (F3L2F) Rabbit Monoclonal Antibody: Antibodies are available* for this critical synaptic priming protein that target both total and cryptic forms, serving as a biomarker readout for TDP43 dysfunction and loss. (*Contact us for pricing and availability.)
- Cryptic HDGF2: HDGF2 cryptic protein is only translated when TDP43 fails to repress cryptic splicing. In order to detect cryptic HDGF2, antibodies with non-overlapping epitopes are necessary.
-
-
Optimized Matched Antibody Pairs: Transitioning to high-throughput immunoassay screens requires capture and detection antibodies with non-overlapping epitopes. CST offers rigorously validated matched antibody pairs that are purpose-built for high-signal, low-noise performance in complex lysates and biofluids:
-
Targeted Immunoassay Kit: The PathScan® TDP43 Sandwich ELISA Kit #16486 provides a ready-to-use solution that detects endogenous levels of TDP43.
Beyond its direct role in RNA regulation, TDP43 pathology often intersects with other classic neurodegenerative proteins, including tau, α-synuclein, and β-amyloid. An emerging target of interest in this network is TMEM106B, a type II transmembrane lysosomal protein genetically linked to TDP43 proteinopathies like FTD and LATE.
While the exact mechanisms are still being uncovered, the relationship between these two proteins appears to converge on lysosomal dysfunction. Under pathological conditions, TMEM106B undergoes sequential proteolysis, generating fragments that accumulate into intra-lysosomal amyloid fibrils. Evidence suggests that this aggregation may compromise lysosomal integrity, leading to impaired TDP43 clearance from the cytoplasm and accelerating toxic seeding. To support further research into TMEM106B biology in TDP43 proteinopathies, CST offers domain- and cleavage-specific antibodies to detect both total and processed forms of TMEM106B.
|
TMEM106B Targets - Total & Cleaved Isoforms: |
|
By providing exclusive tools to assess both TDP43 aggregation and the functional consequences of splicing failure, CST enables scientists to build a more complete, actionable picture of neurodegeneration—enabling hypothesis-driven experimentation and supporting the journey from the bench to preclinical diagnostic and therapeutic opportunities.

REFERENCES
-
Forman MS, Trojanowski JQ, Lee VM. TDP-43: a novel neurodegenerative proteinopathy. Curr Opin Neurobiol. 2007;17(5):548-555. doi:10.1016/j.conb.2007.08.005
- Babazadeh A, Rayner SL, Lee A, Chung RS. TDP-43 as a therapeutic target in neurodegenerative diseases: Focusing on motor neuron disease and frontotemporal dementia. Ageing Res Rev. 2023;92:102085. doi:10.1016/j.arr.2023.102085
- Wu Y, Wanga J, Zhao Q. Advancements in TDP-43 research: Towards biomarkers and therapeutic targets for amyotrophic lateral sclerosis. Aging and Health Research. 2024 Nov;5:100215. doi: 10.1016/j.ahr.2024.100215.
- de Boer EMJ, Orie VK, Williams T, et al. TDP-43 proteinopathies: a new wave of neurodegenerative diseases. J Neurol Neurosurg Psychiatry. Published online November 11, 2020. doi:10.1136/jnnp-2020-322983
- Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79(3):416–438. doi:10.1016/j.neuron.2013.07.033
- Josephs KA, Murray ME, Whitwell JL, et al. Updated TDP-43 in Alzheimer's disease staging scheme. Acta Neuropathol. 2016;131(4):571–585. doi:10.1007/s00401-016-1537-1
- Nasir AR, Delpirou Nouh C. TDP-43-proteinopathy at the crossroads of tauopathy: on copathology and current and prospective biomarkers. Front Cell Neurosci. 2025;19:1671419. Published 2025 Oct 28. doi:10.3389/fncel.2025.1671419
- Brenton JW, Follett J, Nirujogi R, et al. TDP-43 loss of function drives aberrant splicing in Parkinson's disease. Preprint. bioRxiv. 2025;2025.09.04.673943. Published 2025 Sep 8. doi:10.1101/2025.09.04.673943
- McKee AC, Stern RA, Nowinski CJ, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain. 2013;136(Pt 1):43–64. doi:10.1093/brain/aws307
- Cohen TJ, Lee VM, Trojanowski JQ. TDP-43 functions and pathogenic mechanisms implicated in TDP-43 proteinopathies. Trends Mol Med. 2011;17(11):659-667. doi:10.1016/j.molmed.2011.06.004
- Sinha IR, Atkinson AL, Irwin KE, Ling JP, Wong PC. TDP-43: [GU]-ardian of the transcriptome. Mol Neurodegener. Published online May 14, 2026. doi:10.1186/s13024-026-00944-2
- Liu Y, Yan D, Yang L, Chen X, Hu C, Chen M. Stathmin 2 is a potential treatment target for TDP-43 proteinopathy in amyotrophic lateral sclerosis. Transl Neurodegener. 2024;13(1):20. Published 2024 Apr 11. doi:10.1186/s40035-024-00413-0
- Agra Almeida Quadros AR, Li Z, Wang X, et al. Cryptic splicing of stathmin-2 and UNC13A mRNAs is a pathological hallmark of TDP-43-associated Alzheimer's disease. Acta Neuropathol. 2024;147(1):9. Published 2024 Jan 4. doi:10.1007/s00401-023-02655-0
- Irwin KE, Jasin P, Braunstein KE, et al. A fluid biomarker reveals loss of TDP-43 splicing repression in presymptomatic ALS-FTD. Nat Med. 2024;30(2):382-393. doi:10.1038/s41591-023-02788-5
- Ma XR, Prudencio M, Koike Y, et al. TDP-43 represses cryptic exon inclusion in the FTD-ALS gene UNC13A. Nature. 2022;603(7899):124-130. doi:10.1038/s41586-022-04424-7
- Hruska-Plochan M, Wiersma VI, Betz KM, et al. A model of human neural networks reveals NPTX2 pathology in ALS and FTLD. Nature. 2024;626(8001):1073-1083. doi:10.1038/s41586-024-07042-7
- Zhong W, Scialò C, Gatta B, et al. Lysosomal escape and TMEM106B fibrillar core determine TDP-43 seeding outcomes. Preprint. bioRxiv. 2025;2025.12.19.695531. Published 2025 Dec 22. doi:10.64898/2025.12.19.695531