If you’ve performed CUT&RUN, CUT&Tag, or ChIP/ChIP-seq experiments, you know how important it is to use high-quality antibodies to generate data you can trust. However, did you know that CST also offers chromatin profiling assay support reagents that will help ensure data quality and reproducibility?
This blog discusses how non-antibody support reagents can impact your data and answers the following questions you might have about optimizing your chromatin profiling experiments:
- Which beads should I use for my ChIP/ChIP-seq experiment?
- How do I protect chromatin and antibody epitopes when sonicating chromatin for ChIP/ChIP-seq?
- How can I minimize variability when fragmenting chromatin with MNase for ChIP/ChIP-seq?
- Can I use DNA purification columns in my CUT&RUN or CUT&Tag experiment?
- What concentration of digitonin should I use for my CUT&RUN or CUT&Tag experiment?
- How much pAG-MNase and pAG-Tn5 should I use for my CUT&RUN and CUT&Tag experiments?
CST is here to support all of your chromatin profiling needs—developing, formulating, and stringently validating these ancillary reagents before they hit your bench to safeguard your data quality. Bonus? These reagents are available as off-the-shelf CUT&RUN, CUT&Tag, or ChIP/ChIP-seq kits or à la carte for your convenience.
After all, who wants to spend all that time running an experiment just to have it fail or yield ambiguous results because a buffer or enzyme didn’t perform the way it should?
Which beads should I use for my ChIP/ChIP-seq experiment?
Protein G agarose or magnetic beads are used in combination with antibodies to enrich for chromatin-bound proteins of interest. However, there are many types of beads available. How do you know which beads will deliver the best chromatin immunoprecipitation (ChIP) and ChIP-seq data? CST has developed ChIP-grade beads to make your bead selection easier.
CST scientists evaluated different types of beads to identify the ones that provide optimal enrichment and minimal background. Different conjugation chemistries were also evaluated when developing the ChIP-grade magnetic beads. To yield better signal-to-noise ratios, ChIP-Grade Protein G Magnetic Beads #9006 are formulated in a BSA-containing buffer to block the binding of non-specific proteins, while ChIP-Grade Protein G Agarose Beads #9007 are supplied in a buffer that contains both BSA to block non-specific protein binding and sonicated salmon sperm DNA to block non-specific DNA binding.
Just a note that if you’re performing ChIP-seq, you’ll need to use the ChIP-grade magnetic beads since the salmon sperm DNA bound to the agarose beads can contaminate your sequencing results.
ChIP-Grade Beads Yield Better Signal-to-Noise Compared to IP-Grade Beads

Figure 1. ChIP was performed using digested chromatin from HeLa cells and the indicated antibodies, using either ChIP-Grade Protein G Magnetic Beads #9006 or IP-Grade Protein A Magnetic Beads. Purified DNA was analyzed by quantitative real-time qPCR, using primers to the indicated genes. The purified DNA is expressed as a percentage of the total input chromatin.
How do I protect chromatin and antibody epitopes when sonicating chromatin for ChIP/ChIP-seq?
Sonication is one method used to fragment chromatin so it is soluble and readily coprecipitated. However, sonication can be very harsh and can result in damage to both the chromatin and antibody-binding epitopes. Cell and nuclear lysis buffers from CST are formulated to better protect the integrity of the chromatin and antibody epitopes, but are still effective at fragmenting chromatin and making the antibody binding accessible for immunoprecipitation (IP). This is particularly important when investigating transcription factors and cofactors since they bind more weakly to chromatin than histone marks and are often less abundant.
SimpleChIP® Sonication Cell and Nuclear Lysis Buffers #81804, also available as part of the SimpleChIP Plus Sonication Chromatin IP Kit #56383, delivers equivalent next-generation sequencing (NGS) results for histone marks when compared to the competition. However, the NGS signal-to-noise ratios for transcription factors and cofactors indicate that the CST buffers contribute to delivering superior results compared to the competition.
CST Sonication and Cell Nuclear Lysis Buffers Outperform Competitors When Analyzing Transcription Factor or Cofactor Interactions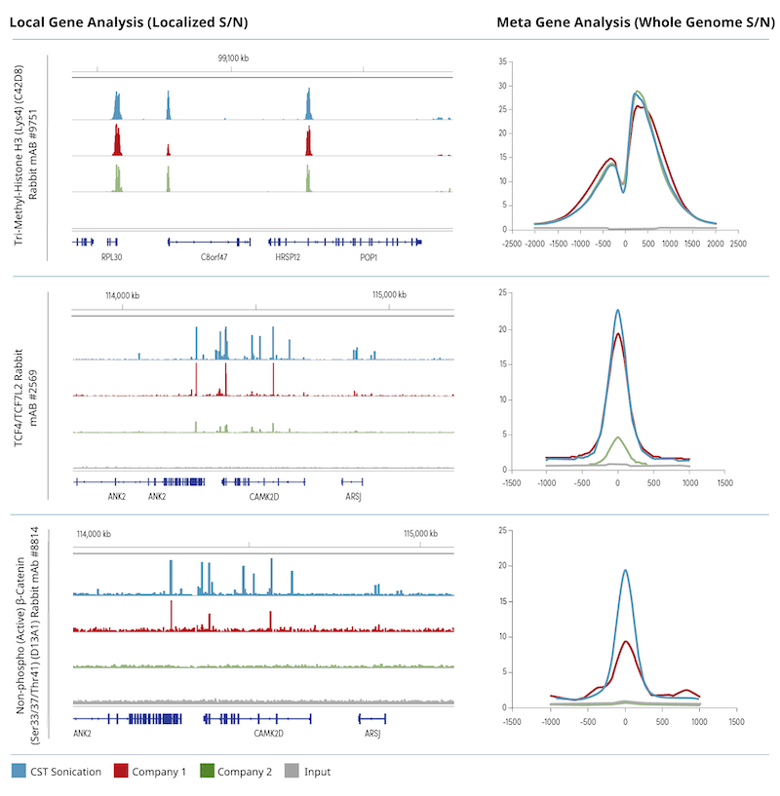
Figure 2. Chromatin was prepared using cross-linked chromatin from 4 x 106 HCT 116 cells according to the protocols provided by the kit manufacturers and immuno-enriched using the indicated antibodies. The ChIP-seq tracks on the left show enrichment of the protein of interest across a localized region of interest. The meta-gene analysis on the right depicts the signal-to-noise ratio of all identified peaks found across the whole genome. Input sample was used as a negative control for both the ChIP-seq track view and meta-gene analysis.

How can I minimize variability when fragmenting chromatin with MNase for ChIP/ChIP-seq?
Another option when fragmenting chromatin is to use micrococcal nuclease (MNase) instead of sonication. Something that needs to be considered when using MNase is that the enzymatic activity can vary between lots, even if used at the same concentration. To help you avoid this source of inter-assay inconsistency and save you time, CST normalizes the Micrococcal Nuclease #10011 for enzymatic activity, not concentration, during the validation process. Titration experiments are performed for each new lot of MNase to evaluate activity side-by-side with a previous lot, so you’ll always get the same chromatin digestion pattern when you add 0.5 µL of MNase from CST to four million cells.
In the example below, the new lot of MNase is formulated to exhibit the same activity as the old lot, both generating the desired chromatin digestion pattern.
Normalizing MNase by Enzymatic Activity Ensures Lot-to-Lot Consistency
Figure 3. Four million HDLM-2 cells per digestion were formaldehyde-crosslinked and chromatin was prepared and digested as described in the SimpleChIP Plus Chromatin Immunoprecipitation Protocol (Magnetic Beads), using increasing amounts of two different lots of Micrococcal Nuclease #10011. Digested chromatin DNA samples were separated on 1% agarose gel as indicated. The desired chromatin digestion banding pattern is shown in lane 4 and lane 9.
Can I use DNA purification columns in my CUT&RUN or CUT&Tag experiment?
CUT&RUN and CUT&Tag are faster and more cost-effective alternate methods to ChIP-seq. The Henikoff lab, who developed the CUT&RUN and CUT&Tag assay, initially recommended using phenol-chloroform extraction to purify DNA because CUT&RUN generates DNA fragments that are smaller than fragments generated by ChIP. Unfortunately, phenol-chloroform is a very messy and time-consuming method. Columns offer more ease-of-use, but not all DNA purification columns are created equally. Scientists at CST have validated that our purification columns efficiently purify the smaller DNA fragments generated in CUT&RUN and CUT&Tag assays.
The DNA Purification Buffer and Spin Columns (ChIP, CUT&RUN, CUT&Tag) #14209 recover DNA fragments that are ≥ 35 bps, ensuring recovery of over 98% of the CUT&RUN DNA fragments since less than 2% of these fragments are smaller than 35 bps. The columns are also compatible with CUT&Tag, as tagmented fragments are at least 70 bps since adapters are ligated to the DNA of interest before purification. The DNA fragment recovery for each new column batch is confirmed by real chromatin profiling tests that guarantee consistent performance for your preferred application.
Comparison of DNA Purification Methods for CUT&RUN Fragments.
Figure 4. (A) A low-range DNA ladder mix (lane 1, unpurified) was purified using either DNA Purification Buffers and Spin Columns (ChIP, CUT&RUN, CUT&Tag) #14209 (lane 2) or phenol-chloroform extraction followed by ethanol precipitation (lane 3) and separated by electrophoresis on a 4% agarose gel. As shown, phenol-chloroform followed by ethanol precipitation efficiently recovers all DNA fragment sizes, while DNA spin columns recover DNA fragments ≥ 35 bp. (B) DNA was purified using phenol-chloroform extraction followed by ethanol precipitation from a CUT&RUN assay performed using TCF4/TCF7L2 (C48H11) Rabbit mAb #2569. The size of the DNA fragments in the library was analyzed using a Bioanalyzer system. The adaptor and barcode sequences added to the library during construction account for 140 bp in fragment length. Therefore, starting 35 bp DNA fragments would be 175 bp in length after library preparation (indicated with a blue vertical line in the figure). As shown, less than 2% of the total CUT&RUN enriched DNA fragments are less than 175 bp (starting length of 35 bp), suggesting that the DNA purification spin columns are sufficient for the capture of > 98% of the total CUT&RUN DNA fragments.
What concentration of digitonin should I use for my CUT&RUN or CUT&Tag experiment?
The digitonin used when performing your CUT&RUN and CUT&Tag experiments needs to permeabilize the cell and nuclear membranes just enough so that enzymes and antibodies can get in, but not so much that everything, including the DNA, can come out, since this will increase the assay background. Like MNase, digitonin activity can also vary from lot to lot, even when the same concentration of digitonin is used. Therefore, titration experiments should be performed to evaluate activity.
CST titrates every new lot of Digitonin Solution #16359 to ensure the same level of activity from lot to lot, leading to time savings for you. Each new lot is tested side-by-side with the previous lot and normalized based on signal strength, not concentration. The optimal concentration for the new lot is then tested against the old lot using NGS to confirm equivalent performance.
Digitonin Titration Ensures Consistent Activity Between Lots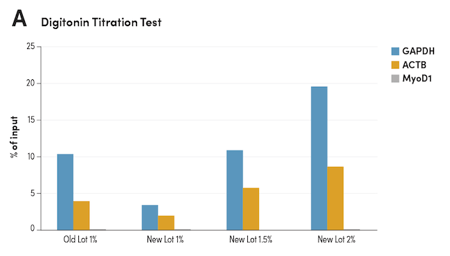

Figure 5. CUT&RUN was performed with HCT 116 cells and Tri-Methyl-Histone H3 (Lys4) (C42D8) Rabbit #9751, using CUT&RUN Assay Kit #86652. (A) Different lots of Digitonin Solution #16359 with various concentrations were used in buffers as indicated. The enriched DNA was quantified by real-time PCR using human GAPDH exon 1 primers, SimpleChIP Human β-Actin Promoter Primers #13653, and SimpleChIP® Human MyoD1 Exon 1 Primers #4490. The amount of enriched DNA in each sample is represented as the signal relative to the total amount of input DNA. (B) The optimal concentration of 1.5% for the new lot Digitonin Solution #16359 was used in buffers and compared to the old lot side-by-side using NGS. DNA Libraries were prepared using DNA Library Prep Kit for Illumina (ChIP-seq, CUT&RUN) #56795. The figures show binding across GAPDH, a known target gene of H3K4me3.
How much pAG-MNase and pAG-Tn5 should I use for my CUT&RUN and CUT&Tag experiment?
CST has developed and formulated pAG-MNase and pAG-Tn5 enzymes for CUT&RUN and CUT&Tag, respectively, for optimal performance and lot-to-lot reproducibility. Each new lot is validated for enzyme activity, not concentration, to ensure consistent results.
The pAG-MNase enzyme used to perform CUT&RUN experiments is tolerant when it comes to the amount used. However, CST still performs side-by-side titrations and NGS tests against the previous lot when formulating and validating the pAG-MNase that comes with the CUT&RUN Assay Kit #86652 and CUT&RUN pAG-MNase and Spike-In DNA #40366.
Normalizing pAG-MNase by Enzymatic Activity Ensures Lot-to-Lot Consistency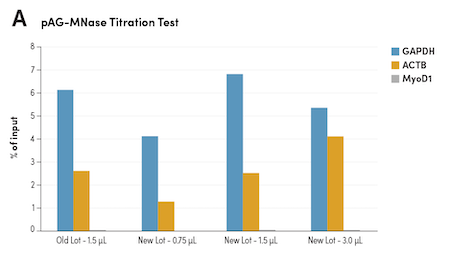

Figure 6. CUT&RUN was performed with HCT 116 cells and Tri-Methyl-Histone H3 (Lys4) (C42D8) Rabbit #9751, using CUT&RUN Assay Kit #86652. (A) Different lots of pAG-MNase #40366 with various volumes were used in the assay as indicated. The enriched DNA was quantified by real-time PCR using human GAPDH exon 1 primers, SimpleChIP Human β-Actin Promoter Primers #13653, and SimpleChIP® Human MyoD1 Exon 1 Primers #4490. The amount of enriched DNA in each sample is represented as the signal relative to the total amount of input DNA. (B) The optimal volume of 1.5 μl of the new lot of pAG-MNase #40366 was used in the assay and compared to the old lot side-by-side using NGS. DNA Libraries were prepared using DNA Library Prep Kit for Illumina (ChIP-seq, CUT&RUN) #56795. The figures show binding across GAPDH, a known target gene of H3K4me3.
A successful CUT&Tag assay is more reliant on the activity of the pAG-Tn5 enzyme than the quantity used, as our in-house tests did not observe an increase in read number, peak number, or signal-to-noise ratio when more than the recommended volume of enzyme was used in each reaction (see more details in the CUT&Tag FAQ page). To ensure consistent lot-to-lot performance, the pAG-Tn5 (Loaded) #79561 enzyme, which also comes with the CUT&Tag Assay Kit #77552, is validated with NGS by titrating the new lot against a previous lot side-by-side. The activity of the pAG-Tn5 enzyme will diminish over time, so it is imperative to store the enzyme at -20°C and to use the enzyme before the expiration date listed on the vial.
Normalizing pAG-Tn5 by Enzymatic Activity Ensures Lot-to-Lot Consistency
Figure 7. CUT&Tag was performed with HCT 116 cells and Tri-Methyl-Histone H3 (Lys4) (C42D8) Rabbit mAb #9751, using CUT&Tag Assay Kit #77552. Different lots of CUT&Tag pAG-Tn5 (Loaded) #79561 with various volumes were used in the assay as indicated. DNA Libraries were prepared using CUT&Tag Dual Index Primers and PCR Master Mix for Illumina Systems #47415. The figures show binding across GAPDH, a known target gene of H3K4me3.
Blog: CUT&Tag DNA Library Yield Too Low to Detect with an Agilent Bioanalyzer or TapeStation System?
Your One-stop Chromatin Profiling Shop
Save yourself time and headache by using ChIP, CUT&RUN, and CUT&Tag reagents developed and validated at CST. Our scientists have spent the time optimizing buffers, enzymes, and DNA purification columns so you get the best results. We also perform titrations for you and compare them against previous lots for lot-to-lot consistency, ensuring reproducibility over time. Couple that with antibodies rigorously validated for your application of choice and you’ll be generating chromatin profiling data you trust before you know it.
23-FLE-89700