In part one, Understanding the Proteome: An Introduction to Mass Spectrometry-Based Proteomics, we gave an overview of mass spectrometry-based proteomics. Now, it’s time to talk about how this strategy can be used to identify peptides with post-translational modifications (PTM) from a complex biological sample.
Antibody Enrichment Explained: Proteomics Is Like Fishing...
It’s a beautiful day out on the lake. The boat is bobbing up and down, your fishing rod is set up, and the fish have been biting all afternoon. Maybe the next one will be a keeper. Wait! Stop daydreaming about fishing! You’re actually at the lab bench late on a Friday afternoon. It’s obvious why you’re thinking about your weekend fishing trip, though—it’s because you’re puzzling over a proteomics experiment and proteomics can be a lot like fishing. (Yes, really!)

Let’s say you’ve digested your cell or tissue sample into a complex mixture of shorter peptides. Remember, the proteome comprises as many as a million proteoforms, which means your mixture contains millions of peptides, some modified and some not. A big challenge when studying PTMs is that modified peptides are generally present at a much lower abundance in a peptide mixture than their unmodified counterparts. Some are extremely rare. So, how do you increase your chances of identifying and quantifying them by mass spectrometry?
The PTM peptides can first be fished out of the protein mixture by antibody enrichment or immunoaffinity purification (IAP). As every fisherman (or woman) knows, to be successful at catching fish, you must use the right bait. So let’s look at what is in your tackle box...
- Standard, site-specific PTM antibody: The antibody recognizes the modified amino acid in the context of a specific sequence of amino acids surrounding it. For example, an antibody to human Akt1 phosphorylated at serine 473 would encompass the sequence FPQFpSYSAS.
- PTM-sequence motif antibody (motif antibody): The antibody recognizes the modified amino acid within a certain motif. For example, substrates of Akt1 generally contain an RXRXXpS motif, where X is any amino acid. The motif antibody will recognize the sequence RXRXXS in the Akt substrate when the serine residue is phosphorylated.
- PTM-specific antibody (PTM-antibody): The antibody will recognize any residues with the PTM of interest. For example, an antibody to acetyl-lysine will recognize all acetylated lysine residues regardless of which amino acids flank them.
What Is PTMScan®?
Using these antibodies to enrich for PTM peptides and then running the enriched peptides in Liquid Chromatography with tandem mass spectrometry (LC-MS/MS) is what CST calls the PTMScan® method. PTMScan allows for the identification and quantification of hundreds to thousands of PTM peptides in a single LC-MS/MS run.
The modification-specific antibodies listed above can be used in different ways depending on the type of question you are trying to answer in your experiment. So, which one should you choose?
PTMScan Direct
If you have an idea of which signaling pathway is involved in the system you are studying, you can perform what CST calls PTMScan Direct. In this version of PTMScan, mixes of standard site-specific antibodies are made and immobilized on beads. The antibody mix contains standard site-specific antibodies that target critical regulatory sites on proteins within a signaling pathway, for example, proteins involved in Akt/PI3K signaling. The site-specific antibodies immunoprecipitate the corresponding PTM-containing peptides out of the peptide mixture, and the purified peptides are then identified and quantified by LC-MS/MS. PTMScan Direct allows a detailed view of the regulation of known signaling pathways.
PTMScan Discovery
Another approach to IAP for mass spectrometry-based proteomics is what CST calls PTMScan Discovery. This is similar to PTMScan Direct in that it uses immobilized antibodies to enrich PTM peptides from a mixture. Unlike PTMScan Direct, which uses standard site-specific antibodies, in PTMScan Discovery, each IAP bead has only one type of antibody immobilized to it, and the antibody is a PTM-specific or motif-specific antibody. This approach allows capture of any peptide that contains the PTM or PTM-motif of interest, such as substrates of a particular kinase, acetylated peptides, or ubiquitinated peptides.
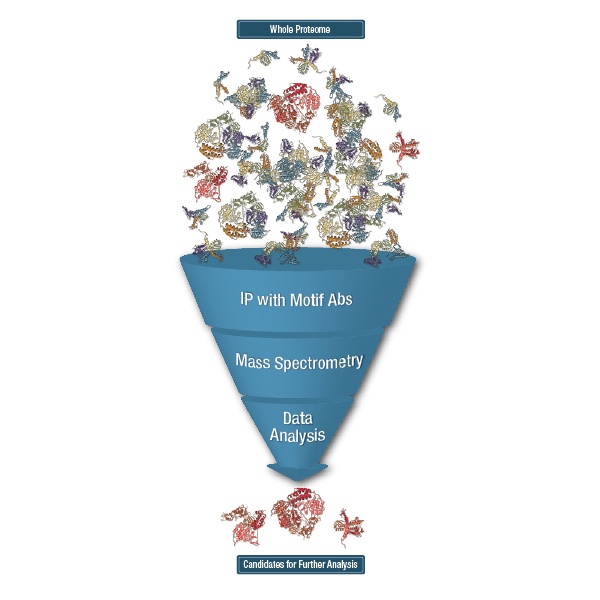
Comparing IMAC to Antibody-Based Enrichment
Another strategy for purifying phosphorylated peptides specifically before LC-MS/MS is called immobilized metal affinity chromatography (IMAC). IMAC takes advantage of inherent coordination chemistry by using immobilized metal ions, commonly Fe3+, to capture phospho-peptides, which are negatively charged. To go back to our fishing analogy, compared to PTMScan, IMAC is more like fishing with a net. It is an effective way to purify phospho-peptides, but it is less targeted than PTMScan Discovery and tends to favor the more abundant peptides in the mixture. IMAC is a highly complementary technique to PTMScan.
PTMScan Discovery and IMAC, followed by LC-MS/MS, can both lead to the identification and quantification of hundreds to thousands of PTM-containing peptides. The trick then is to assess which are the most relevant candidates for follow-up. A good first step is determining which peptides have the biggest difference in abundance between the various samples run, for example, between treated cells and untreated cells or cancer tissue and healthy tissue. From there, context can be given to the proteomic data using previous knowledge of expected biology or use of pathway analysis tools. The best candidates can then be validated by western blots, IP-westerns, siRNA or overexpression studies, site-directed mutagenesis, or other assays to provide actionable candidates for future research.
So you can see, by using these techniques, fishing for peptides can be as rewarding as a day out on the lake, and they will help move your research forward quickly so you can get out on the lake sooner.
Up Next in the Proteomics Series...
The last blog post in this series, From Discovery to Diagnostics: Translational Applications of PTM Proteomics, features a bench-to-bedside story as an example of how PTMScan can be applied.
To learn more, watch the "Post-translational Modification: Antibody Enrichment for Mass Spectrometry-based Proteomics" webinar.