After sequencing of the human genome was complete, it was time to roll up our sleeves and get started on the daunting task of unraveling the complexity of the proteome. Thus, the era of proteomics, the study of the function of all expressed proteins, was born. This task is especially complicated because, unlike the human genome, which is largely static in every cell, the proteome is different between, for example, a liver cell and a brain cell, or between a healthy cell and a cancerous cell, or even between an individual cell at the different stages of development.
To address this challenge, the Human Proteome Project was founded. Its mission is to characterize all human genes by generating a map of the protein-based molecular architecture of the body. In this way, it has become a resource to help elucidate the biological and molecular function of genes and facilitate the advanced diagnosis and treatment of disease.
What Are Post-Translational Modifications (PTMs)?
The 20,000 or so identified genes are translated into as many as a million protein forms, or proteoforms. So why is there such a disparity between the number of genes and the number of proteoforms? Alternative promoters and gene splicing contribute to some of the proteome’s complexity, but a large part is due to post-translational modification (PTM) of proteins. PTMs are covalent modifications that include the addition of phosphoryl, acetyl, methyl, ubiquityl, or other groups to specific amino acids, as well as proteolytic cleavage. These modifications are essential for protein activity, subcellular localization, degradation, and protein-protein interactions. Understanding the role of PTMs in disease states is key to identifying biomarkers and developing therapeutics.
Bottom-Up vs Top-Down Approaches
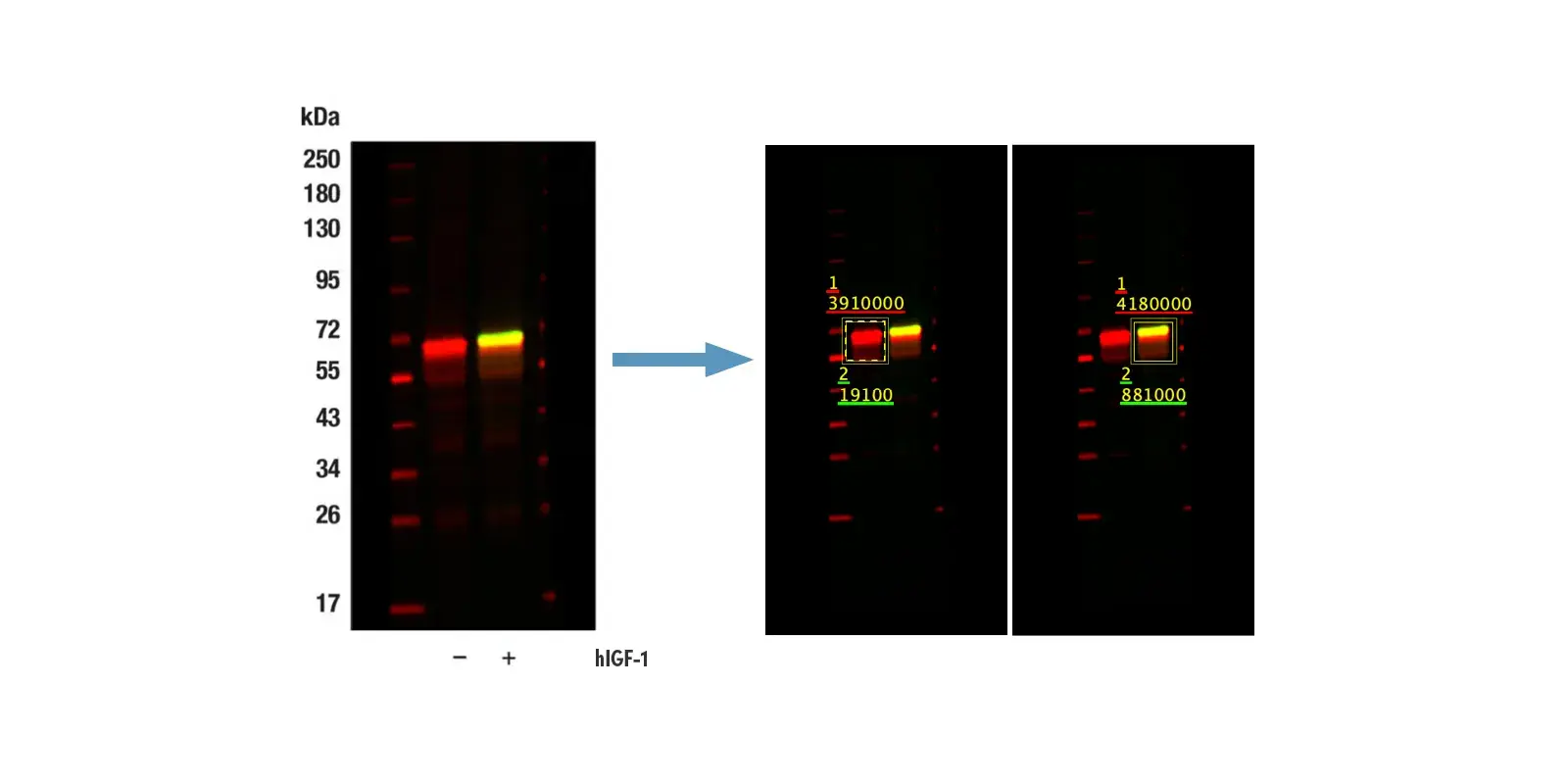
Mass spectrometry (MS) is a highly sensitive technology used to study proteins and their PTMs in a sample on a large scale. Advances in mass spectrometry technology have given proteomics a boost. The most established and widely used strategy is called bottom-up proteomics, where the cell/tissue sample is digested with a protease, often trypsin, into short peptides before being subjected to MS for identification. The characteristics of the proteoform are then inferred from the peptide information.
Top-down proteomics, in which the sample is not digested prior to mass spectrometry, is less widely used, but has potential application in clinical research due to the additional information that can be garnered from analysis of the native protein (degradation products, sequence variations, and combinations of PTMs).
There are many different types of MS. A common one used for proteomics is liquid chromatography tandem MS, or LC-MS/MS. In LC-MS/MS, digested peptides are separated by high-performance liquid chromatography before being introduced to the mass spectrometer. Exquisitely accurate mass-to-charge ratio measurements of the intact peptide (MS1) and fragments of the same peptide (MS2 or MS/MS) are collected by the instrument. These mass-to-charge measurements are matched to a database of all proteins, allowing identification of the peptide.
Mass spectrometry-based proteomics is a powerful tool for determining novel substrates of phosphorylation, ubiquitination, acetylation, etc.; identification and validation of drug targets; discovery of biomarkers; elucidation of off-target drug effects; and exploration of the mechanism of action of drugs/chemical modulators.
Learn More
In the blog in our Introduction to Proteomics series, Enriching for PTMs: Antibody-Based Strategies for Proteomics Analysis, we will feature two bottom-up approaches for identifying and quantifying PTMs in complex biological samples.
You may also want to watch the Simplifying Proteomics webinar.