In recent years, immune checkpoints in the tumor microenvironment have been under intense study. If you work in the immuno-oncology field, chances are you are either performing multiplex immunohistochemistry (mIHC) or would like to. mIHC enables the detection of the spatial location of six or more biomarkers—including immune checkpoint proteins—in formalin-fixed, paraffin-embedded (FFPE) tissue samples, and it is critical when cataloging subsets of immune infiltrate and cancer cells and their interactions in the tumor microenvironment.
Ultimately, a multiplexed image like the one featured here provides a multi-layered depiction of a tumor, with each antibody corresponding to a different fluorescent signal.

Multiplex IHC analysis of breast carcinoma tissue probed with a 6-plex panel using PD-1 #86163 (green), TIM3 #45208 (yellow), LAG3 #15372 (magenta), CD8α #70306 (orange), and Pan-keratin #4545 (cyan), plus DAPI to label nuclei (blue).
If you want to detect more targets in your IHC but aren’t sure how to design a panel of antibodies and fluorophores for mIHC, we’ll walk you through the process in this post, paying special attention to antibody order as it relates to epitope masking.
Multiplex IHC for Characterizing the Tumor Microenvironment
Visualizing tumors with mIHC gives you the ability to characterize the immune infiltrate and test hypotheses in novel ways. For example, Are T cells localized primarily to the tumor periphery, along the invasive margin or within the tumor itself? Are those that are present expressing markers of T cell exhaustion? How close in proximity are PD-1+/CD8+ T cells to PD-L1+ macrophages? Are the CD8+ T cells in the tumor activated?
Questions like these are becoming critical to the understanding of response rates to PD-1 blockade and the movement past PD-L1 as the main biomarker.
Antibodies for Multiplex IHC Panel Design
When designing an mIHC antibody panel, there are many factors to consider. A tyramide-based multiplex approach may be the simplest way to achieve strong signal in FFPE tissue. If you have the tissue, IHC-validated antibodies, and tyramide-fluorophore conjugates at hand, you’ll be able to set up a protocol. One of the biggest factors to consider is how much of each antibody you will need, as less antibody is often required when detecting with tyramide compared to a chromogen such as DAB (3,3′-Diaminobenzidine).
Once you get a sense for the relative intensity of the staining for each antibody, you then need to figure out the best way to achieve a balanced set of signals in your final panel. Ideally, you want to pair the antibodies with the strongest labeling and/or the most abundant target expression with the weakest available tyramide-fluorophore conjugates, and pair the weakest-staining antibody with the brightest conjugates.
Primary Antibody Stripping & Potential Effects on Epitopes
The mIHC protocol involves multiple heating cycles in order to remove (strip) the primary and secondary antibodies in between each staining round. This introduces another set of factors to take into account when fitting all the pieces of the puzzle together. While some epitopes may be largely unaffected by this heating, others are more heat-sensitive and will degrade with each round of heating, demonstrating progressively weaker signal. On the other hand, other epitopes can become more exposed with each additional heating step and will demonstrate stronger signal as each cycle is performed. Antibodies to epitopes that fall into this last category will optimally be placed in one of the last slots in the panel.
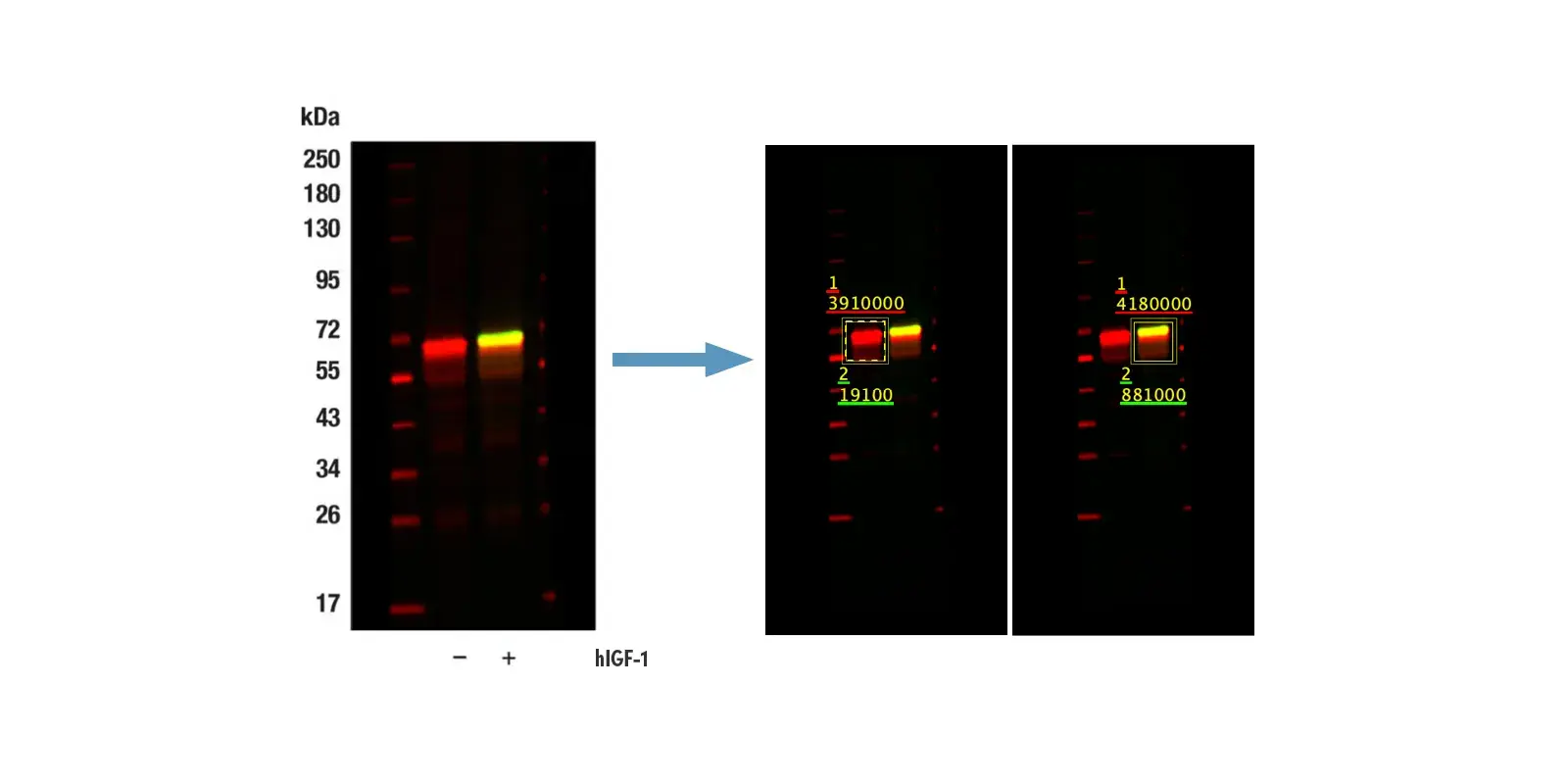
You should also consider the efficiency of primary antibody stripping. To test for efficient stripping, you can perform a normal staining round with steps for primary, secondary, tyramide detection, and heating. Then, perform a second round, this time omitting the primary antibody, with the same secondary and tyramide amplification steps but substituting a second fluorophore for the tyramide conjugate. Low staining in this channel indicates complete stripping, as in the example below (left panel). If you observe staining in the second channel, as shown in the CD4 test below (right panel, compare signal in the red channel), it's an indication that stripping of the primary antibody is incomplete, and you should further optimize conditions to ensure complete removal of the primary.

Multiplexed IHC analysis of mouse glioblastoma tumor tissue using PD-1 #84651 (green, left) or CD4 #25229 (cyan, right). To test stripping efficiency, secondary-only incubation was subsequently performed as described in the text using SignalStain Boost IHC Detection Reagent (HRP, Rabbit) #8114 and detected with Cy5 tyramide conjugate (red).
How to Get Around Epitope Masking When Multiplexing
A phenomenon called epitope masking is another factor that can add complexity to achieving accurate staining when multiplexing. Epitope masking can occur when you have multiple antibodies in a panel that are directed against epitopes located in the same subcellular compartments of the same cells in a tissue. As tyramide is deposited over the first such epitope in the panel, it can blanket nearby epitopes that you are attempting to stain in later slots of the panel. In the below example, the absence of overlap between CD3 and CD8 in a panel examining the immune infiltrate of mouse syngeneic tumors caught our attention.

Multiplexed IHC analysis of 4T1 lung metastasis (mouse syngeneic tumor) using recombinant monoclonal antibodies CD3ε (D4V8L) Rabbit Monoclonal Antibody #99940 (green) and CD8α (D4W2Z) Rabbit Monoclonal Antibody (Mouse Specific) #98941 (red). Blue = DAPI. The CD8 antibody was applied first.
As shown above, the red CD8+ T cells are largely exclusive of the green CD3+ T cell in the field. While these tumor infiltrates can include a subset of CD8+/CD3- cells, such as dendritic cells and NK cells, we wouldn’t expect the majority of the CD8+ cells in a given field to fall into this category. As CD3 and CD8 are targets that we would expect to stain the same tiny membranes of the same tiny T cells, epitope masking was a likely explanation to account for these observations.
There are a few ways to approach epitope masking. Often, switching the order in which these epitopes are stained will be sufficient to resolve the issue. This was the case for our CD8/CD3 example. As you can see below, once CD3 was placed first in the ordering, the expected amount of overlap between these two targets was observed.
 Multiplexed IHC analysis of 4T1 lung metastasis (mouse syngeneic tumor) using recombinant monoclonal antibodies CD3ε (D4V8L) Rabbit Monoclonal Antibody #99940 (green) and CD8α (D4W2Z) Rabbit Monoclonal Antibody (Mouse Specific) #98941 (red). Blue = DAPI. The CD3ε antibody was applied first.
Multiplexed IHC analysis of 4T1 lung metastasis (mouse syngeneic tumor) using recombinant monoclonal antibodies CD3ε (D4V8L) Rabbit Monoclonal Antibody #99940 (green) and CD8α (D4W2Z) Rabbit Monoclonal Antibody (Mouse Specific) #98941 (red). Blue = DAPI. The CD3ε antibody was applied first.
The expression levels of certain proteins can vary widely from others, and the higher expression of one can possibly make it less likely to be significantly masked by deposited tyramide than another. The epitope composition itself, namely the amount of tyrosine it contains, may also make it more or less likely to be masked by tyramide deposited in a previous round.
In the event that switching antibody order does not sufficiently alleviate the epitope masking problem, other approaches can be used to achieve a better balance between targets in successive steps. These approaches can include titration of the secondary antibody, titration of the tyramide-fluorophore complexes themselves, or optimization of incubation time (secondary and/or tyramide-fluorophore complexes).
Ultimately, while it can be easy to spot epitope masking when you have near-complete absence of a marker expected to be present on certain cell types, it can be more difficult to recognize in other cases. Markers of T cell exhaustion, for example, also co-localize to the membranes of the same T cells but aren’t necessarily always present, as the presence of exhaustion and the level of exhaustion can vary between tumors.
 Multiplex IHC analysis of non-small cell lung carcinoma probed with a 6-plex panel for T cell exhaustion proteins, plus DAPI to label nuclei. Multiplexed image of all seven channels (upper left) is shown with a region of interest (white box in upper left) from which the individual channels are displayed.
Multiplex IHC analysis of non-small cell lung carcinoma probed with a 6-plex panel for T cell exhaustion proteins, plus DAPI to label nuclei. Multiplexed image of all seven channels (upper left) is shown with a region of interest (white box in upper left) from which the individual channels are displayed.
Panel Optimization: Comparing Multiplex and Single-Plex
In order to ensure that epitope masking or other factors aren’t decreasing your staining levels, we recommend that you stain single-plex fluorescent slides in parallel with a multiplexed slide prepared using the theoretically optimal conditions. Before considering your panel fully optimized, make sure that there aren’t significant increases or decreases in the numbers of cells staining positively for each target in your multiplexed slide vs your single-plex slides. This last step will ensure that not only is epitope masking caught early on, but that the level of signal you observe for each multiplexed target is an accurate reflection of the biology in your samples.
Multiplex IHC Solutions for Spatial Biology Research
CST offers an expansive IHC-validated portfolio of unconjugated and conjugated antibodies for interrogating the TME using antibodies to key phenotypic and functional biomarkers, including PD1, CD25, TIM3, CD4, LAG3, CD8, and many more.
The SignalStar® multiplex IHC technology is ideal for visualizing up to eight phenotypic and functional biomarkers in the same panel. The technology uses antibodies, oligonucleotides, and fluorophores to interrogate cellular presence, location, function, and biomarker co-expression patterns. Learn more about the SignalStar multiplex IHC technology on the resource page, or download the app note below.

CST Solutions for Multiplex Spatial Biology
Visit the Multiplex Spatial Biology Resource Center to explore the full suite of CST offerings. Fuel your spatial biology research with our diverse, validated portfolio of low- to high-plex solutions compatible with the sample types, platforms, and workflows used in multiplex spatial biology and imaging.