Senescence is a cellular state during which cells remain metabolically active, but irreversibly withdraw from the cell cycle and fail to respond to proliferation-inducing stimuli. Senescent cells influence a number of physiological and pathological processes from cancer to diabetes and aging. Accordingly, understanding why senescence contributes to these conditions may lead to the development of pro- and anti-senescence therapies to treat a range of diseases.
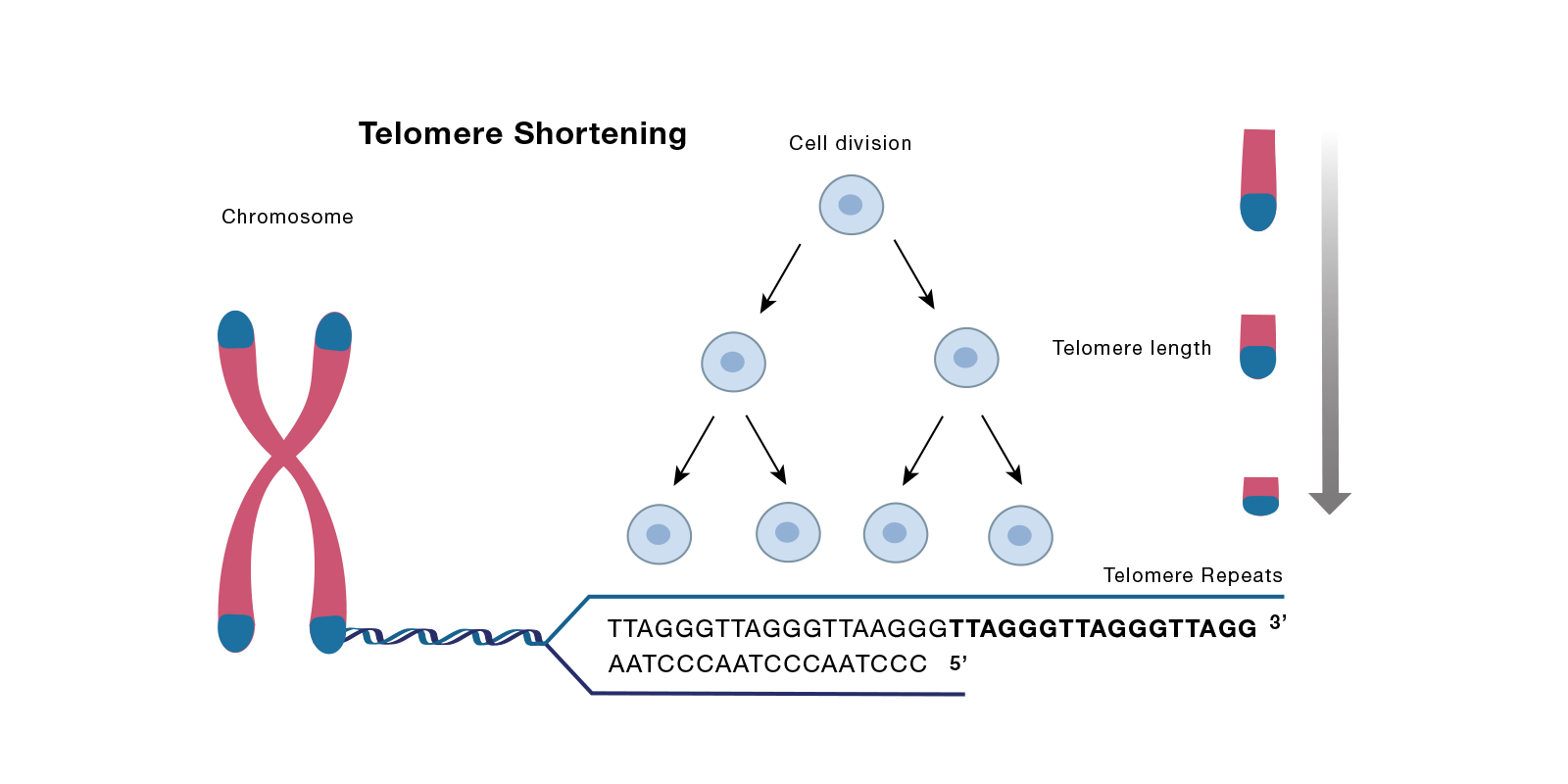
DNA damage through various extracellular signals and intracellular oncogenic activation are primary initiators of cellular senescence. However, senescence can also arise as the result normal shortening of telomeric DNA. Decades ago, Leonard Hayflick and Paul Moorehead reported their seminal discovery that cells grown in vitro have a finite capacity to undergo successive rounds of cell division before they undergo permanent cell cycle arrest – a process termed replicative senescence (Hayflick and Moorhead, 1961; PMID: 13905658). Subsequent work has shown that this limit is imposed in part due to signals derived from the trimming of the ends of chromosomes– called telomeres– that occurs with each successive round of DNA duplication prior to cell division [(Bodnar AG, 1998; PMID 9454332) or the following review (Kuilman T 2010, PMID 21078816)] .
Telomeres are composed of hundreds to thousands of short repeating nucleotide sequences, along with a single-stranded GT-rich overhang. These sequences are recognized and bound by several telomere-binding proteins, known as the shelterin complex, that aid in the formation of the chromosome cap to protect the ends of chromosomes from degradation. The shelterin complex is composed of several proteins including telomere repeat factor 2 (TRF2), telomeric repeat-binding factor 2-interacting protein (TERF2IP, also known as RAP1), and telomere protection protein (TPP1). With each round of cell division the length of telomeric sequences shortens due to the fact that chromosomes are linear strands of DNA whose ends cannot be fully copied during replication, a phenomenon termed the end replication problem. When these sequences reach a critical length, the cellular DNA damage repair machinery is activated which, in turn, causes the cell to undergo senescence. To counteract this process, cells express an enzyme called telomerase which serves to restore telomeric sequences. However, with aging and in certain cell types where telomerase activity is low, replicative senescence ensues.
Many types of cancer cells exhibit unrestricted proliferative capacity and escape replicative senescence in part through the upregulation of telomerase expression and activity. Telomerase acts in concert with a protein complex which includes dyskerin (DKC1) to synthesize telomeric end sequences de novo during cell division. The Mre11-Rad50-Nbs1 (MRN) complex also plays an important role in maintaining telomere stability and mutations in genes that encode this complex are associated with an increased risk of some cancers.
Senescent cells accumulate in tissues during aging primarily through replicative senescence caused by telomere shortening. Accordingly, drugs that selectively target senescent cells or increase telomerase activity to limit replicative senescence are of potential therapeutic value as they may reverse the deleterious effects of aging and extend lifespan in humans. In fact, since telomeres function as a cellular clock, methods to slow their degradation may prolong tissue health and function by limiting cellular senescence.
Cells that have undergone replicative senescence exhibit many of the morphological and metabolic characteristics associated with senescence, which results in a response to other forms of DNA damage. Accordingly, assays to detect changes in the expression of proteins that regulate cell cycle arrest and the adoption of the senescence-associated secretory phenotype (SASP) can be used to detect its presence in a variety of experimental models.
For more information on telomeres and their relationship to cellular senescence please visit the CST Senescence Signaling pathway.