You might be surprised to learn that diabetes, a metabolic disease, may be linked to the neurodegenerative condition Alzheimer’s disease (AD). Diabetes is driven by altered insulin signaling from either insufficient insulin production (Type 1) or altered insulin receptor (IR) signaling (Type 2). In a seemingly unrelated disease, the human e4 allele of APOE is the strongest genetic risk factor for AD.


How are diabetes and Alzheimer’s disease connected?
APOE encodes human apolipoprotein E (ApoE), which is a lipid carrier essential for cholesterol homeostasis. Natural allele variations result in the expression of three ApoE isoforms: E2, E3, and E4. Even small differences in the amino acid sequence of ApoE can significantly increase the risk for AD, with APOE4 allele carriers at the highest risk compared to the more common APOE3 allele. As it turns out, APOE4 carriers exhibit altered brain insulin signaling and glucose metabolism, suggesting a mechanistic clue as to how diabetes and AD may be linked.
In a remarkable study published in Neuron, Zhao et al. sought to investigate the potential mechanistic link between the Alzheimer's risk gene APOE4 and altered metabolism. The researchers used an APOE mouse model in which the murine Apoe gene was replaced with human APOE3 or APOE4. By examining aged brain lysates from each genotype, the authors observed age-dependent changes in basal insulin signaling. That is, APOE4-aged mice exhibited decreased IR signaling compared to APOE3 mice when measuring the phosphorylation states of downstream IR substrates, Akt (Ser473) and GSK-3β (Ser9). Based on these findings, the authors hypothesized that ApoE may directly alter insulin signaling in an isoform-specific manner.
Consistent with their hypothesis, the authors observed ApoE isoform-dependent alterations in insulin-dependent cellular signaling: APOE4 mice exhibited reduced insulin-stimulated phospho-Akt compared to APOE3 mice. Similar results were also seen in primary neurons derived from Apoe -/- mice when recombinant ApoE3 or ApoE4 was directly applied to cultures, with or without insulin. This suggests that the ApoE protein may directly modulate insulin signaling. How might this be the case?
The authors examined the possibility that ApoE might bind directly to the insulin receptor (IR). Using recombinant protein and ELISA assays, the authors observed direct ApoE binding to IR. Remarkably, ApoE4 exhibited enhanced IR binding compared to ApoE3.
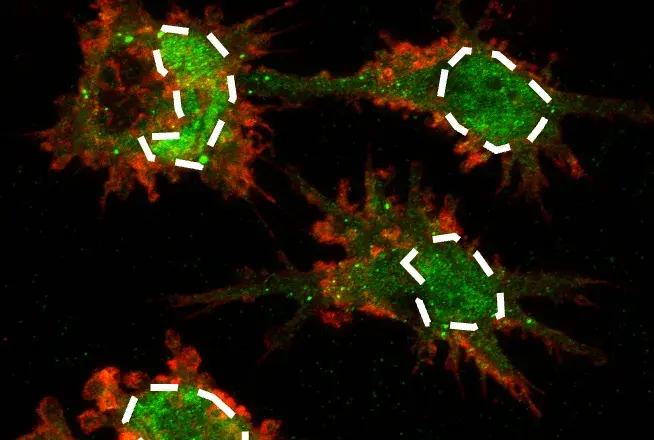
How can ApoE4-dependent reduction in downstream IR signaling and enhanced ApoE4/IR interaction be mechanistically reconciled? IR cell surface availability is regulated, in part, by circulating levels of insulin. Interestingly, ApoE4 treatment led to an overall reduction of cell surface IR. Moreover, ApoE4 acted synergistically with insulin to drive a greater reduction of cell surface IR than co-treatment with ApoE3. Importantly, the authors also observed ApoE4-specific enrichment with IR in early endosomes, at least in cultured cells, suggesting a mechanism by which ApoE-specific isoforms might trap IRs in the cellular trafficking machinery to alter IR signaling.
ApoE4 and the Insulin Receptor — It's a Trap!
This study proposes a novel mechanism by which specific Alzheimer’s disease-linked ApoE isoforms might impair insulin signaling in the brain, potentially contributing to disease progression. However, several questions remain. For example, the authors found that ApoE4-dependent changes in IR signaling and its effect on IR stability accelerate with age. Are these alterations correlated in time with altered behavior or cellular alterations (e.g., synaptic dysfunction) observed in AD mouse models? A shared timeline might suggest a common pathway to disease.
Furthermore, are there mechanistic differences between “classic” (i.e., acute) insulin-mediated signaling endosomes that activate IR pathways and chronic ApoE/insulin-mediated IR endosomal entrapment that reduces IR signaling pathways?
Finally, could some brain cells be more sensitive to altered insulin receptor signaling than others? Certainly, more research into the convergence of these seemingly disparate pathways will elucidate a way forward to understanding the mechanistic trap that is AD.
Insulin Receptor Signaling and Alzheimer's Disease Signaling Pathways from CST