In November 2017, over 30,000 neuroscientists gathered in Washington D.C. to talk all things brain at the Society for Neuroscience (SfN) meeting. I’ve attended SfN for what is now on the order of decades. Having reluctantly accepted veteran status for the annual meeting, I thought this year’s SfN was an opportunity to consider where neuroscience has been, where it is, and where it’s going.
The research objectives at SfN — to understand how molecules, cells, and circuits drive complex behavior — are broad and overwhelming. Even today, I choke up recalling that inevitable question I’d get from my graduate school advisor after a meeting — what did you learn? Such a question is challenging for any meeting, and a strong perspective, particularly for SfN, is necessary to even consider it.
SfN 2017: Neurons Are Not Alone
From my own perspective, I have always taken a more “bottom-up” approach. My own research interests lie in synapses, the singular unit of neuronal information transfer in the brain. During grad school and my postdoc, I investigated how molecules mediate synapse formation, function, and plasticity (change in efficacy) to better understand synaptic contributions to neuronal circuits and, ultimately, behavior (i.e., a bottom-up approach). Synapses are fundamentally cellular interactions between neurons that, with a reductionist approach, can be studied in the absence of other cell types that exist in the brain.
But neurons are not alone. In the brain, neurons function in the complex context of a milieu of non-neuronal cells that greatly outnumber neurons. I recall a remarkable poster presentation at the 2005 SfN meeting regarding mechanisms of synaptic scaling, a homeostatic form of neuronal plasticity. The mechanism, reported by Robert Malenka’s group at Stanford, involved the trafficking of AMPA-type glutamate receptors, which in and of itself was not particularly remarkable to me. However, the authors reported finding that a non-neuronal cell supplied TNF-α as a key molecular mediator of synaptic plasticity represented something of a moment of clarity. At that meeting, I more fully recognized that non-neuronal brain cells contribute to fundamental neuronal processes, like synaptic plasticity, in ways that we did not fully appreciate or understand.
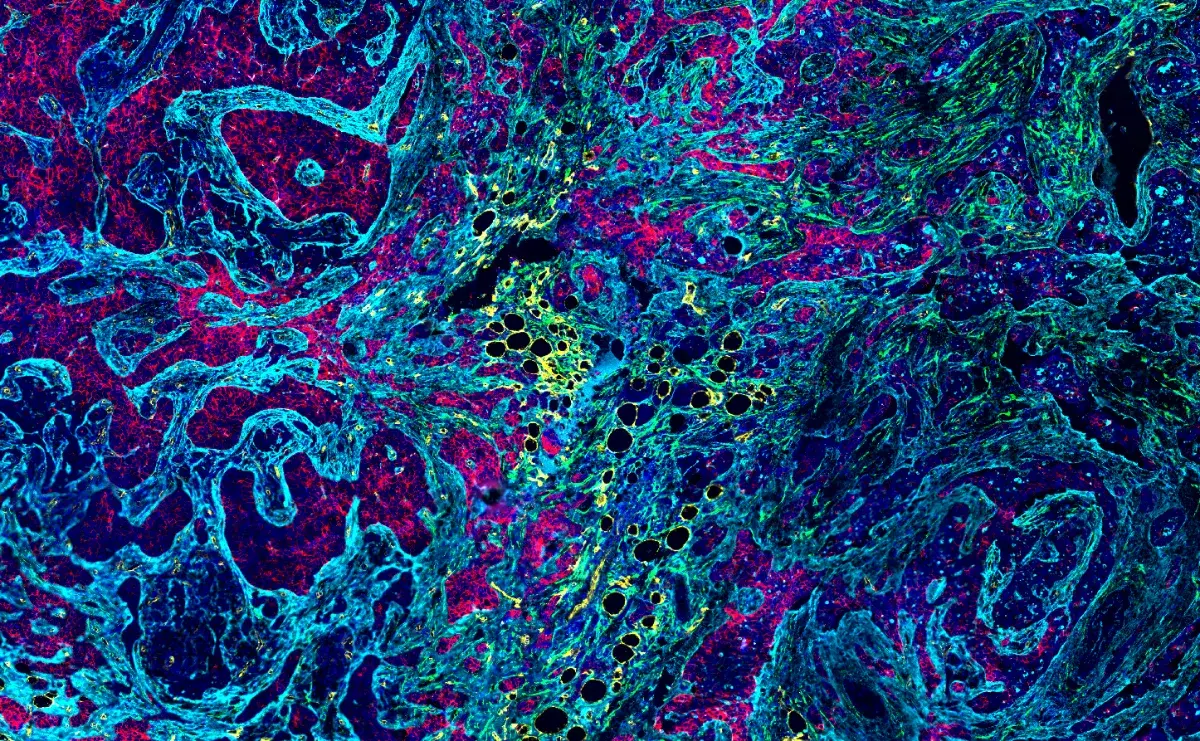
Fast-forward almost 10 years and non-neuronal cells are now all the rage at SfN, both in the context of normal brain function and disease. Several of this year’s posters/talks captured my attention regarding non-neuronal cells — especially microglia — as they relate to Alzheimer’s disease (AD). AD is a poorly understood neurodegenerative disease associated with amyloid plaques and neurofibrillary tangles. Investigators are trying to understand how microglia, resident macrophages of the brain, contribute to AD neuronal loss and dysfunction. The most basic of questions (e.g. are they protective or detrimental?) are at play.
Why are microglia on the radar for so many investigators? Microglia are associated with amyloid plaques, the pathological hallmark of AD. Moreover, several microglia-specific genes are associated with AD risk. Finally, new technologies are emerging to study microglia and their functions.
 A few of the 30,000(!) attendees at SfN 2017.
A few of the 30,000(!) attendees at SfN 2017.
So where are we at right now? Here are a few research highlights that captured my imagination at the 2017 SfN meeting:
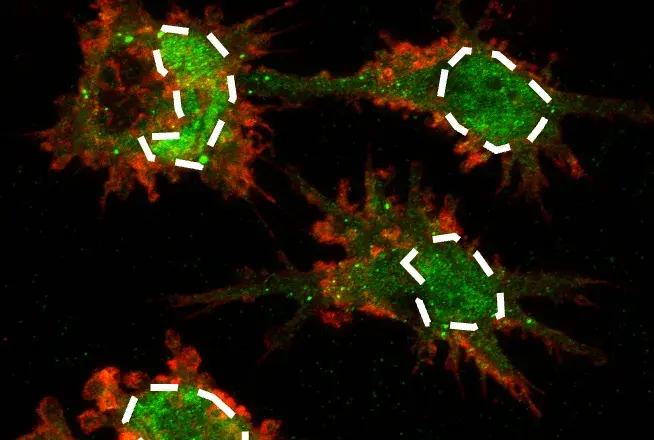
At the “Alzheimer’s Disease and Neuroinflammation” nanosymposium, Orit Matcovitch-Natan described her work in Ido Amit’s laboratory at the Weizmann Institute of Science identifying and classifying specific microglial transcriptomes associated with specific disease conditions. Microglia are dynamic cells that, like peripheral macrophages, are “activated” under certain conditions. Matcovitch-Natan utilized a single-cell RNA-seq approach to compare the transcriptional profiles of microglia derived from control and AD model (5xFAD) mice, and reported the identification of a transcriptionally unique microglia type (DAM) present in the AD mice. Consistent with a direct relationship between AD and microglia, several of the identified markers are uniquely upregulated in microglia surrounding the amyloid plaques. The upregulation of these DAM markers in microglia surrounding amyloid plaques suggest an interaction that is capable of initiating and/or sustaining the DAM transcriptional profile. The finding may also suggest that specific pathways that may be activated to mobilize specific DAM functions. The Amit laboratory proposed at least 2 specific DAM-activated states, suggesting a complex underlying biology that needs further investigation. Future work will likely reveal additional DAM states. Are these states more temporally and spatially defined, and are there disease-specific DAMs?
In the same nanosymposium, Sarah Hopp from Brad Hyman’s laboratory at Massachusetts General Hospital described a novel cellular mechanism by which pathological tau could spread from neuron to neuron. In addition to amyloid plaques, AD patients exhibit neurofibrillary tangles that result from misfolding of the microtubule-associated protein tau. Tau, of course, is an intracellular protein that is known to spread from neuron to neuron. The specific transport mechanism pathological tau employs for interneuronal transport remains a mystery, but Hopp described findings suggesting microglia may be involved. Using microglia isolated from human AD patients and a tauopathy AD mouse model (rTg4510), Hopps reported that microglia contain fragments of tau, likely a product of microglial clearance of neurofibrillary tangle-containing neurons. Intriguingly, these fragments of tau serve as tau “seeds” to promote misfolding of normal tau, at least in vitro. How might microglia contribute to the spreading of these seeds? Hopp provided evidence that microglia package these seeds into exosomes that are taken up by normal neurons. This novel mechanism of interneuronal transport of pathological tau by microglia has several implications. Could tau-containing exosome formation be hijacked to prevent spreading of the disease? Could microglia function be enhanced/altered to prevent generation/spreading of tau seeds? Do microglia-associated genetic risk factors contribute to this process? Future work will likely bring clarity to these pressing questions.
During the poster session on “Neuroinflammation and Alzheimer’s Disease,” Michael Audrain from Sam Gandy and Michelle Ehrlich’s groups at the Icahn School of Medicine described the convergence of an AD-associated microglia signaling pathway with a process that involves removal of synapses. Microglia are known to sculpt neuronal connections through a process called synaptic pruning. Synaptic pruning occurs during development, but can also occur in adults under normal and disease conditions. The complement protein, C1q, is likely the so-called “eat me” signal that identifies synapses to be pruned by microglial phagocytosis. Mutations in TREM2, a membrane microglial protein that regulates many microglia processes including phagocytosis, are linked to increased AD risk. The Gandy lab reported that modulating Dap12 levels, a downstream effector of TREM2, was sufficient to modulate the synaptic C1q pruning phenotype in mouse AD models. Interestingly, Brian Leung from Terrence Town’s laboratory at the Keck School of Medicine, University of Southern California independently presented findings at the “Protective and Pathogenetic Mechanisms in Alzheiemer’s Disease” nanosymposium, reporting a direct interaction between TREM2, C1q, and Aβ monomers (the building blocks of amyloid plaques). Together, these two studies suggest an emerging active area of research where molecules from neurons and non-neuronal cells, like microglia, converge at the cellular level to contribute to AD progression.
SfN 2017 and Beyond
What will be the big thing at next year’s SfN and beyond? Of course, that is hard to say, but I suspect the challenge ahead is to more deeply understand how neurons integrate with non-neuronal cells (astrocytes and microglia) to drive normal and disease processes. So many questions remain. For example, if C1q is an “eat me” signal, might a “don’t eat me” signal exist? How many microglial activation states exist, and what tools need to be developed to interrogate and identify these cellular states? How can microglia, astrocytes, and neurons be effectively studied both in vivo and in vitro that accounts for the temporal and spatial complexities of the brain? Investigation into how this integration works and how they might be altered in disease will be critical, potentially revealing neurodegenerative disease mechanisms and novel therapeutic targets.