It's almost time for the 2017 Society for Neuroscience meeting. To get your neurons excited for the meeting, here's a journal club discussing a recent paper with interesting findings for Alzheimer's disease.
The pathological hallmark of Alzheimer’s disease (AD) is the accumulation of amyloid β (Aβ) plaques and neurofibrillary tangles. Despite decades of research, the direct (and indirect) contribution of these lesions in disease progression is poorly understood. Do these lesions directly cause neuronal dysfunction and neurodegeneration? If so, why do some patients accumulate these lesions, but exhibit normal neurological behavioral before death? Do AD patients have secondary defects in cellular and/or molecular processes that normally function to protect patients from accumulation of these nefarious lesions? If so, what are these cell types and what…
Wait.
Take a breath.
You get the point. This is the type of rabbit hole that researchers are well familiar with. Without falling into that hole ourselves, let’s get some context. Until recently, investigators focused most of their efforts on the neuron, the cellular node through which information is transmitted in the brain. But in the context of the brain, how do other cells contribute to AD? We’ve long known that microglia, resident macrophages of the brain, surround amyloid plaques of AD patients, but their specific role in AD progression remains unclear. We can go down another rabbit hole of questions, but is there a molecular stepping-stone that could put us on firmer ground?
TREM2
Recently, investigators identified Trem2 (Triggering Receptor Expressed on Myeloid cells 2) loss of function mutations that associate with AD susceptibility. Importantly, TREM2, a single pass membrane protein, is expressed uniquely in microglia. Last year, Yuan et al. reported TREM2-enriched microglia and their processes surround Aβ plaques, suggesting a TREM2-dependent mechanism exists to mobilize microglia around these lesions (1). Consistent with this hypothesis, the authors found that genetic manipulation of Trem2 results in decreased formation of microglia surrounding Aβ lesions, leading to more diffuse, rather than compact, plaques. Therefore, the authors proposed that microglia serve as neuroprotective microglia barriers to surround and, perhaps, protect neurons from pathological Aβ lesions in a TREM2-dependent manner.
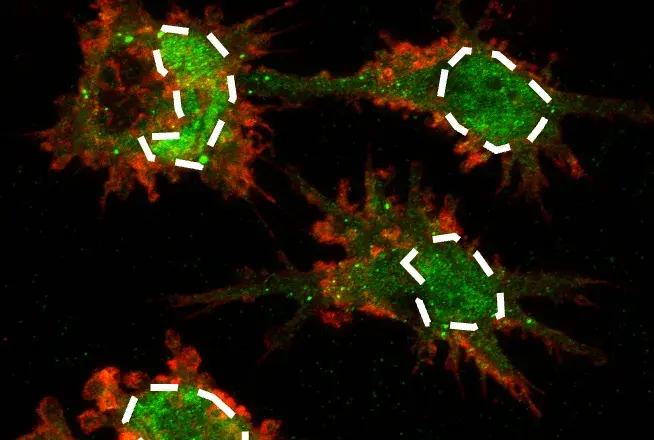
Recent work from Marco Colonna’s group at Washington University provides further mechanistic insight (2). Using subcellular resolution microscopy, the Colonna group observed large multivesicular and multilamellar structures in Aβ plaque-associated microglia from AD mouse model tissue, but only in the absence of functional TREM2. The appearance of these structures is consistent with enhanced autophagy, a “self-eating” process to maintain cellular metabolism under stressful conditions.
Several molecular hallmarks of autophagy exist. Using antibodies to these markers, Colonna’s group described the molecular recruitment of autophagocytic pathways in microglia surrounding plaques, but only in the absence of functional TREM2, both in human patient samples and AD mouse models. Based on these observations, the Colonna group hypothesized that TREM2 normally functions to maintain normal cellular metabolism under high-energy processes, like when microglia surround plaques.
Connection to Autophagy
If that’s true, does TREM2 mobilize normal metabolic pathways under normal conditions? The mammalian target of rapamycin (mTOR) pathway regulates autophagy, and many tools exist to interrogate this important pathway. Using bone marrow-derived macrophages (BMDM) as a model for microglia, the Colonna group observed defective mTOR activation in BMDMs lacking TREM2, suggesting TREM2 maintains mTOR-dependent signaling pathways under both normal conditions and during processes that require ramped up metabolism. Altering this pathway results in enhanced autophagy. Remarkably, bypassing TREM2 to restore these metabolic pathways, by mobilizing a TREM2-independent upstream pathway (Dectin-1) or by downstream ATP supplementation, was sufficient to ameliorate enhanced autophagocytosis in the absence of TREM2, at least in vitro. Can manipulations of these pathways correct altered TREM2-dependent microglia function in AD?
To address this question, the Colonna group used genetically manipulated AD mouse models lacking TREM2. They found that diet-supplemention of cyclocreatine, a creatine derivative that can enhance regeneration of metabolic ATP, ameliorates microglia autophagy in TREM2-deficient AD mice. Finally, the enhanced accumulation of dystrophic neurites observed in TREM-2 deficient AD mice was reduced when the mice were diet-supplemented with cyclocreatine. These results suggest that microglia normally function to surround and contain pathological AD plaques in a TREM2-dependent manner, and that in the absence of functional TREM2, altered microglial metabolism may contribute to AD progression.
Together, the Colonna group’s findings significantly furthers our understanding of how microglia could act in the normal brain to keep pathogenic plaques at bay, and how microglia dysfunction, via TREM2, may indirectly contribute to AD and other neurodegenerative diseases. Several questions remain, however, in that it is not entirely clear how TREM2 is directly mediating these microglial functions. Could TREM2 be the receptor for Aβ? If so, are they receptive only to Aβ plaques and not other forms of Aβ? How can microglial metabolism be therapeutically targeted to maintain optimal function, especially in patients harboring TREM2 mutations?
Future work will likely shed some light to these pressing questions. Speaking of shedding, did you know TREM2 mutations result in enhanced TREM2 shedding from the cell surface? If so, what is the function of secreted TREM2 and how does this…?
Another rabbit hole for another time.
Interested in autophagy signaling? Explore the autophagy signaling pathway.
References
- Yuan P, Condello C, Keene CD, Wang Y, Bird TD, Paul SM, Luo W, Colonna M, Baddeley D, Grutzendler J (2016) TREM2 haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy.Neuron 90(4), 724-739.
- Ulland TK, Song WM, Huang SC, Ulrich JD, Sergushichev A, Beatty WL, Loboda AA, Zhou Y, Cairns NJ, Kambal A, Loginicheva E, Gilfillan S, Cella M, Virgin HW, Unanue ER, Wang Y, Artyomov MN, Holtzman DM, Colonna M. (2017) TREM2 Maintains Microglial Metabolic Fitness in Alzheimer's Disease.Cell 170(4):649-663.e13.