Although the rise of cancer immunotherapy may seem meteoric to many, the history actually stretches back over a century, when early clinicians noticed something unexpected: In a subset of patients, severe infections coincided with the regression of advanced tumors.
This foundational discovery is attributed to William B. Coley, often called the father of immunotherapy. In the 1890s, he noted a link between tumor regression and the severe skin infection erysipelas and, based on this observation, went on to treat patients with inoperable sarcoma or carcinoma using live bacteria.¹ Building on this early recognition of immune–tumor interactions, in 1909, Paul Ehrlich proposed the cancer immune surveillance hypothesis, suggesting that cancers arise spontaneously but that tumors can be recognized and repressed by the immune system.2
Why Modern Checkpoint Blockade Falls Short
Despite this long history, however, it is only in recent years that immunotherapy has become a central pillar of cancer treatment, largely due to the clinical success of PD-1 and PD‑L1 blockade. These therapies can induce durable remissions with relatively limited immune‑related adverse events (irAEs) compared with earlier approaches such as IL‑2, which proved too toxic at the doses required to achieve tumor regression. The key difference is that PD‑1/PD‑L1 blockade targets a specific tumor‑driven immune evasion mechanism exploited by tumors to dampen T cell activity, whereas IL‑2 broadly amplifies immune activation and toxicity together.
However, only a small percentage of patients respond to PD‑1/PD‑L1 inhibitors, underscoring the fact that tumors use multiple, complex, and often overlapping immunosuppressive mechanisms to evade immune detection and attack. Improving therapeutic success while minimizing toxicity will require a clearer understanding of which immunosuppressive mechanisms dominate in each individual tumor.
The Cellular Requirements for Immune Response
A successful response to PD-1/PD-L1 blockade requires more than just drug exposure. Tumor-specific T cells must be present, able to detect tumor antigens, and capable of executing effector functions such as cytotoxicity. These T cells also depend on dendritic cells for activation and antigen presentation. However, both T cells and dendritic cells can be physically excluded from the tumor, limiting their ability to interact with malignant cells.4
Even when T cells do effectively infiltrate the tumor, they frequently co‑express other inhibitory receptors, such as TIM‑3 and LAG3, so PD‑1/PD‑L1 blockade alone may not be sufficient to restore full effector function.5
Finally, the T cell needs to become activated within the tumor microenvironment. However, the TME often contains high levels of immunosuppressive molecules and cell types such as regulatory T cells, myeloid‑derived suppressor cells, and suppressive “M2‑like” macrophages, all of which can blunt antitumor immunity despite checkpoint inhibition.

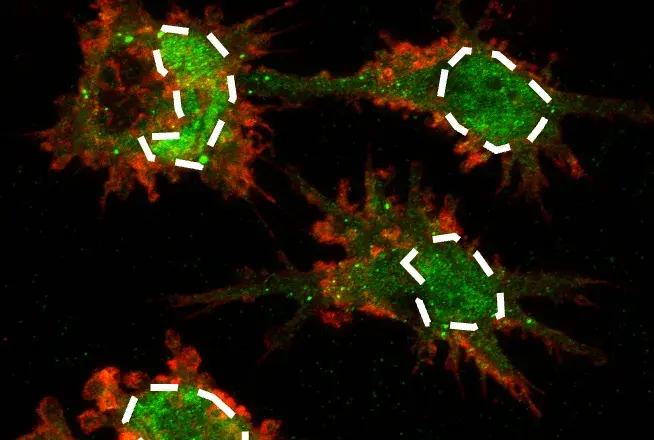
SignalStar® multiplex immunohistochemical analysis of paraffin-embedded human non-small cell lung carcinoma using TIM-3 (D5D5R™) & CO-0010-647 SignalStar Oligo-Antibody Pair #15231 (red), CD3ε (D7A6E™) & CO-0001-488 SignalStar™ Oligo-Antibody Pair #92856 (green), Vimentin (D21H3) & CO-0012-594 SignalStar™ Oligo-Antibody Pair #16471 (yellow), TCF1/TCF7 (C63D9) & CO-0006-750 SignalStar™ Oligo-Antibody Pair #53114 (cyan), and ProLong Gold Antifade Reagent with DAPI #8961 (blue). All fluorophores have been assigned a pseudocolor, as indicated. Staining was performed on the BOND RX by Leica Biosystems.
Multiplex IHC: Why In‑Tissue Immune Phenotyping Is Essential
Given this complexity, increasing the success of cancer immunotherapy while limiting toxicity will likely depend on mapping which immunosuppressive mechanisms are active in each tumor. One powerful way to gain this insight is to characterize both the phenotype and function of immune cells directly in the tumor microenvironment.
Traditional immune phenotyping has historically relied heavily on flow cytometry, but tumor immunology introduces specific challenges: FFPE tumor tissue is often the most available sample type, and spatial relationships between immune and tumor cells carry critical information.
For example, whether CD8+ T cells penetrate tumors or remain trapped in stromal regions can offer important clues about response potential. Multiplex IHC enables the simultaneous detection of multiple markers in intact FFPE tissue, making it well-suited for assessing cell identity, activation state, and spatial context in a single assay—provided panels are carefully designed around a limited number of markers.
Immune Cell Phenotyping Marker Guide for IHC
To support translational cancer researchers and immunologists exploring this field, the Immune Cell Marker Guide is a practical phenotyping resource for designing a panel to identify tumor‑infiltrating immune cells in FFPE tissue. The guide focuses on immune cell types known to be most influential in the tumor immune response, including:
- CD8+ T cells involved in direct tumor cell killing
- Dendritic cells with strong cross‑presentation capacity for priming CD8+ T cells
- Immunosuppressive populations such as regulatory T cells, myeloid‑derived suppressor cells, and M2‑like macrophages
Because multiplex IHC is constrained by the number of markers that can be used simultaneously, the guide emphasizes a minimal set of well‑established markers that reliably identify each cell type. In addition to lineage markers, it includes those that can be used to infer cell function—for example, markers associated with cytotoxicity, exhaustion, or regulatory activity—so that researchers can distinguish effector from suppressive or dysfunctional states rather than just counting cells.
Anchoring studies around a consistent set of markers can help facilitate more reproducible data generation and easier comparison of immune profiles across cohorts, time points, or treatment arms.
From Phenotype to Therapeutic Opportunity
By combining phenotype and functional markers in intact tumor tissue, the Immune Cell Marker Guide helps uncover which immunosuppressive pathways may be preventing a productive antitumor response in a given sample. This information can illuminate potential points of therapeutic intervention, whether by combining checkpoint inhibitors, targeting specific suppressive cell populations, or modulating antigen presentation.
Explore IHC-validated antibodies for phenotype markers, functional markers, and immunotherapy targets from CST.
Within the broader toolbox for tumor microenvironment analysis, multiplex IHC–based phenotyping occupies a complementary niche alongside other experimental approaches such as spatial transcriptomics, flow cytometry, and single-cell sequencing. The same markers outlined in the guide can inform a common base panel across platforms—helping researchers to align their tissue‑based assays, single‑cell datasets, and spatial omics readouts around a shared view of key immune cell populations and functions.
Download the immune cell marker guide to:
- Select a focused marker set for multiplex IHC panels when tissue or channels are limited.
- Distinguish key effector and suppressive immune populations within the tumor microenvironment.
- Generate more interpretable, reproducible datasets for studies involving PD‑1/PD‑L1 or other immunotherapies.

CST Solutions for Multiplex Spatial Biology
Visit the Multiplex Spatial Biology Resource Center to explore the full suite of CST offerings. Fuel your spatial biology research with our diverse, validated portfolio of low- to high-plex solutions compatible with the sample types, platforms, and workflows used in multiplex spatial biology and imaging.
Related Blogs
-
Unlock the Missing Marker: Three Ways to Expand Your Multiplex IHC Panel
- Immunophenotyping Using Flow Cytometry or IHC
- Hallmarks of Cancer: Avoiding Immune Destruction
- What are lymphoid cells and how are they identified?
- What are myeloid cells and how are they identified?
Select References
- Coley WB. The Treatment of Inoperable Sarcoma by Bacterial Toxins (the Mixed Toxins of the Streptococcus erysipelas and the Bacillus prodigiosus). Proc R Soc Med. 1910;3(Surg Sect):1-48. doi:10.1177/003591571000301601
- Ribatti D. The concept of immune surveillance against tumors. The first theories. Oncotarget. 2017;8(4):7175-7180. doi:10.18632/oncotarget.12739
- Sanmamed MF, Chen L. A Paradigm Shift in Cancer Immunotherapy: From Enhancement to Normalization. Cell. 2018;175(2):313-326. doi:10.1016/j.cell.2018.09.035
- Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature. 2015;523(7559):231-235. doi:10.1038/nature14404
- Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010;207(10):2187-2194. doi:10.1084/jem.20100643
This blog was originally published in February 2019. An updated version was published in January 2026.