Protein kinases play a vital role in the regulation of nearly all cellular activities. Through phosphorylation, these signaling enzymes are akin to maestros, orchestrating cellular growth, division, and metabolism by modulating proteins (and other kinases) to adjust their activity, regulate their stability, change their subcellular location, or mark them for destruction.
It should come as no surprise, therefore, that dysregulation of these important regulatory proteins is a common mechanism of disease, specifically in cancers, neurological disorders, and developmental conditions, among others. However, while we know of the existence of over 530 kinases in the human genome, over 400 of which are catalytically active, it is often challenging—or nearly impossible—to determine which kinases regulate which target proteins (or corresponding kinase substrate).
It's long been known that the ability to easily determine the kinases responsible for the dysregulation of critical substrates could reveal many novel drug targets and transform the therapeutic landscape—but up until recently, this simply wasn’t possible on a larger scale.
Now, researchers from MIT, Yale School of Medicine, and Weill Cornell Medical College have leveraged powerful computational tools, purified kinases and peptide libraries, and data available on the Cell Signaling Technology resource, PhosphositePlus, to develop an algorithm to predict the kinase responsible for modulating a particular protein substrate.
The tool, The Kinase Library, is now available through PhoshoSitePlus.
A paper titled An atlas of substrate specificities for the human serine/threonine kinome that details the project was published in January 2023 in Nature and focuses on Ser/Thr kinase activation states in cancer.1 Here, you can learn about the groundbreaking work that contributed to this project, as well as how the partnership with CST to create The Kinase Library will help researchers uncover novel drug targets.
A Lifelong Pursuit: Elucidating Protein Kinases in Signaling Pathways
Professor Lewis “Lew” Cantley of Harvard Medical School and Dana Farber Institute, who was a professor at Weill Cornell Medicine during the time of this research, has been interested in kinase signaling for decades. Well known for his groundbreaking discoveries in signaling and metabolism, including the identification of PI3 kinase, Professor Cantley’s work has had a major impact on our understanding of cancer biology and disease progression.
For decades, proteomics researchers like Professor Cantley have used mass spectrometry (MS) to identify proteins that are more highly phosphorylated in cancer cells versus healthy cells. However, MS-based phosphoproteomics only provides information about phosphorylated proteins and does not identify the direct kinases responsible for those phosphorylation events. Additionally, interrogating the MS data to identify potential phosphorylation sites often returns anywhere from hundreds to tens of thousands of possible posttranslational modification (PTM) sites or motifs—magnitudes larger than could be useful to study individually to confirm kinase specificity. In fact, tens of thousands of sites of serine and threonine phosphorylation have been identified on human proteins, with several thousand known to be associated with human diseases and biological processes—but the kinases responsible for these modifications have only been identified for less than 4% of Ser/Thr motifs.
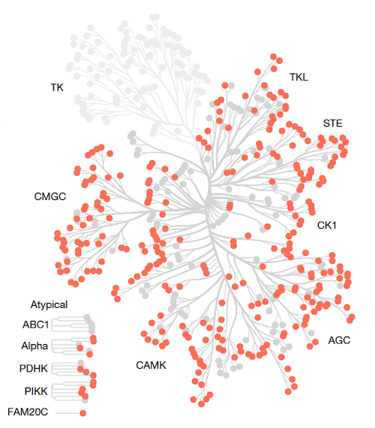
Dendrogram of the human protein kinome, highlighting the Ser/Thr kinases analysed in the study1.
However, modern computational tools are being exploited to provide insights that have not been possible using traditional techniques. With access to the right data, Professor Cantley recognized that a predictive algorithm could be developed that would leverage the structure of the phosphorylation site on the protein to predict which known kinase would be most likely to interact with that protein. But to create such an algorithm, the team first needed to determine the binding preferences for the majority of known human kinases to build their predictive model.
Purifying Ser/Thr Kinases to Determine Binding Preference
This monumental undertaking began over 25 years ago in Professor Cantley’s lab, where he worked with the Nature paper’s other senior authors, Michael Yaffe, director of the MIT Center for Precision Cancer Medicine, and Benjamin Turk, associate professor of pharmacology at Yale School of Medicine. For decades, the trio has been studying kinases and innovating new ways to elucidate their signaling pathways.
One of the trio’s first breakthroughs was the development of a peptide library created to identify the motifs that various kinases interact with. This motif library consists of about 2.5 billion peptide substrates, which represent all the possible amino acid sequences of a peptide motif. To better illustrate this using phosphorylation as an example, the premise is this: The peptide library is constructed by fixing a single phosphopeptide within an amino acid sequence, then varying the amino acids around that fixed phosphopeptide. The result is a "library" that includes all of the possible motifs with which a kinase could interact. Next, the peptide library is subjected to in vitro phosphorylation assays with the kinase of interest and radioactively labeled ATP (gamma P32). The extent of phosphorylation in these peptide arrays provides precise information about which of the 20 amino acids the kinase favors or disfavors in the surrounding positions of its target serine, threonine, or tyrosine site.
Jared Johnson, an instructor in pharmacology at Weill Cornell Medical College, and Tomer Yaron, a former graduate student at Weill Cornell Medical College, are lead authors of the Nature paper and were responsible for determining the substrate binding preferences for the kinases included in the study. The team used Professor Cantley’s peptide library and prioritized determining the binding preferences of the 303 known Ser/Thr kinases, which make up 84% of the kinases predicted to be active in humans.
This included acquiring recombinant preparations of each of the kinases and profiling their activity in their peptide arrays. Using this method, the team could observe which of the substrates the kinase had a stronger preference to phosphorylate based on the surrounding amino acid sequence at their phosphorylation site.
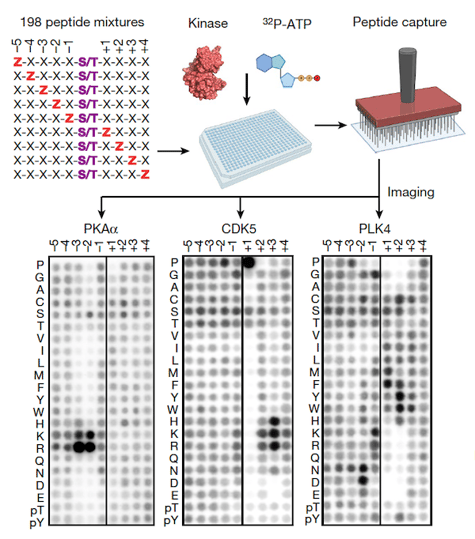 Profiling the substrate specificity of the human serine/threonine kinome. Experimental workflow for the PSPA analysis and representative results. Z denotes fixed positions containing one of the 20 natural amino acids, or either phosphorylated Thr (pThr) or phosphorylated Tyr (pTyr). X denotes unfixed positions containing randomized mixtures of all natural amino acids except Ser, Thr, and Cys. Darker spots indicate preferred residues. Schematic was created using BioRender1.
Profiling the substrate specificity of the human serine/threonine kinome. Experimental workflow for the PSPA analysis and representative results. Z denotes fixed positions containing one of the 20 natural amino acids, or either phosphorylated Thr (pThr) or phosphorylated Tyr (pTyr). X denotes unfixed positions containing randomized mixtures of all natural amino acids except Ser, Thr, and Cys. Darker spots indicate preferred residues. Schematic was created using BioRender1.
Armed with this data about each of the known Ser/Thr kinases, a computational model was developed that could predict kinase substrate binding preference. To score each kinase-peptide pair, the researchers applied their large dataset, normalized and scaled by probabilities, to develop a scoring metric that can be applied to any possible substrate amino acid sequence. They calculated the preference score as a percentile for each kinase-peptide pair by dividing the score between the kinase and given substrate peptide by a background phosphoproteome containing tens of thousands of sites. The preference score represents the degree to which a given kinase prefers a particular substrate compared to all other potential substrates in the cell.
“A Renaissance for Phosphoproteomics”
To the surprise of the researchers, they found many cases where protein kinases with very different amino acid sequences phosphorylate proteins with similar substrate motifs. Additionally, the team found that approximately half of the Ser/Thr kinases studied target one of three major classes of motifs. The remaining half are specific to about a dozen smaller classes of motifs.
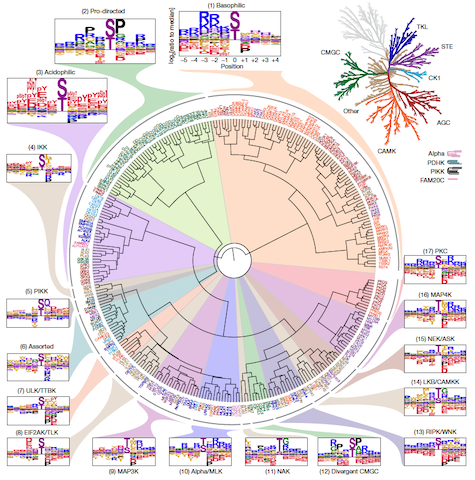
Phosphorylation-site motif tree of the human Ser/Thr kinome. Hierarchical clustering of 303 Ser/Thr kinases on the basis of their amino acid motif selectivity (PSSMs). Kinase names are colour labelled according to their phylogenetic relationships (top right)2. Read the scientific paper to see a full-sized image1.
To test their model, the team applied their prediction system to published untargeted phosphoproteomics datasets. In one example, they analyzed data from cells treated with an anticancer kinase inhibitor called Plk1, which regulates cell growth. Based only on the amino acid sequences of the phosphorylation sites identified and without knowledge of the underlying biology, the team could identify Plk1 as the most frequently predicted kinase in downregulated phosphorylation events. Furthermore, they were able to uncover more distant downstream pathways involving additional groups of kinases, highlighting how their prediction system can organize complicated global phosphoproteomics data into signaling pathways.
The computational model developed by the team is now hosted on Phosphosite in The Kinase Library, where researchers can upload the sequence of the motif site or sites they are studying and receive a list of the kinases that would be most likely to interact with that site.
“With this tool, you can look at the phosphorylated proteins and begin to connect which kinase may have been responsible for that phosphorylation event,” explains Sean Beausoleil, Executive Director of Research at CST.
The tool can be used to interrogate dysfunctional signaling pathways that drive disease, including cancer development and other disorders. After uploading data about the phosphorylation events that change due to a certain treatment, the tool can automate determining the kinases responsible for an increase or decrease in phosphorylation.
“It’s really a renaissance for phosphoproteomics,” continues Sean. “Everyone that has done a phosphorylation experiment should run their data set through this tool.”
Additional Resources
Learn about PTMScan, CST’s product line of proteomics antibodies and kits for identifying PTMs, which were developed in tandem with Professor Cantley using the 2.5 billion peptide library described above.
Select References
- Johnson JL, Yaron TM, Huntsman EM, et al. An atlas of substrate specificities for the human serine/threonine kinome. Nature. 2023;613(7945):759-766. doi:10.1038/s41586-022-05575-3. CC BY 4.0.
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298(5600):1912-1934. doi:10.1126/science.1075762
- Massachusetts Institute of Technology. Enzyme 'atlas' helps researchers decipher cellular pathways: Biologists have mapped out more than 300 protein kinases and their targets, which they hope could yield new leads for cancer drugs. ScienceDaily. ScienceDaily, 12 January 2023.
- Nita-Lazar A, Saito-Benz H, White FM. Quantitative phosphoproteomics by mass spectrometry: past, present, and future. Proteomics. 2008;8(21):4433-4443. doi:10.1002/pmic.200800231
- Han X, Aslanian A, Yates JR 3rd. Mass spectrometry for proteomics. Curr Opin Chem Biol. 2008;12(5):483-490. doi:10.1016/j.cbpa.2008.07.024