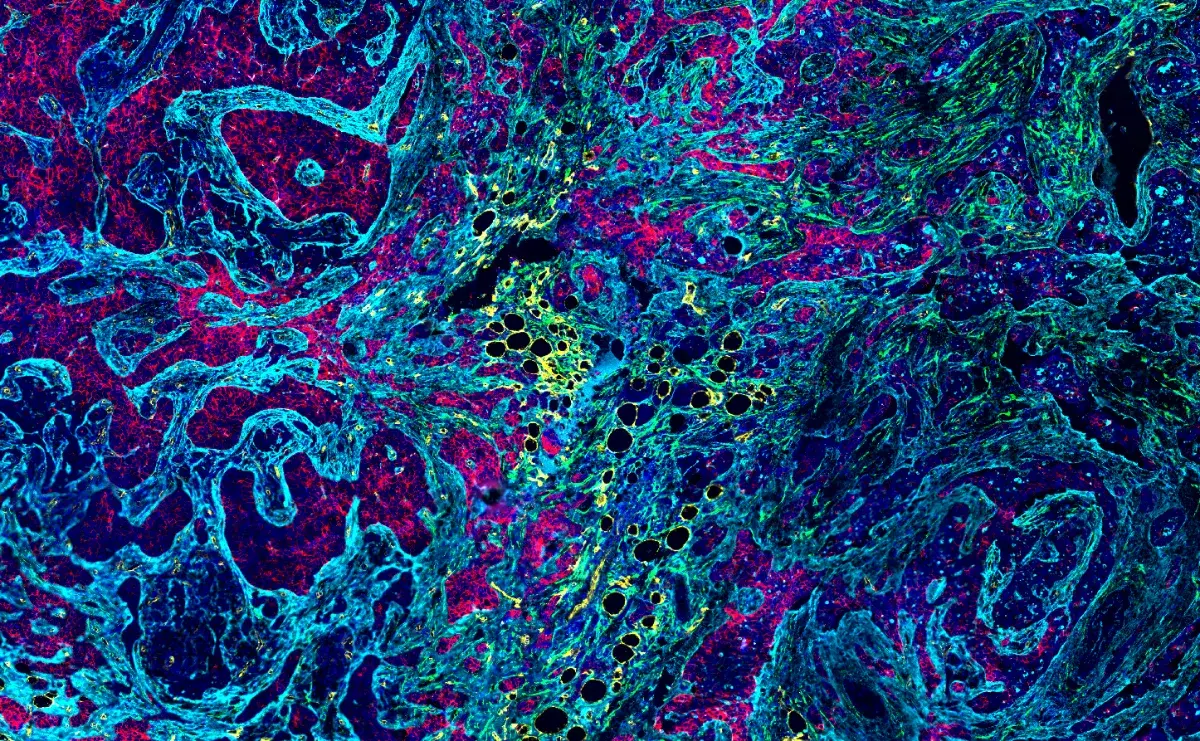
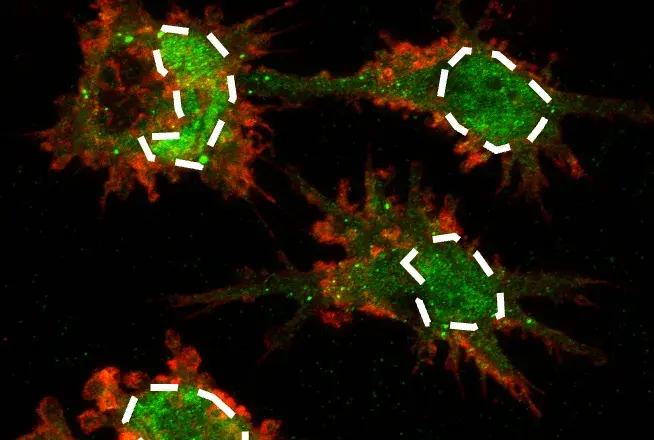
How do cells dynamically regulate access to their tightly packed DNA to control gene expression and respond to environmental cues? The answer lies in chromatin remodeling—a process at the heart of both normal development and cancer. Among ATP-dependent chromatin remodelers, the BAF (short for Brahma-associated factor or BRG1/BRM-associated factor) complexes have emerged as critical regulators, with mutations in their subunits now recognized as major drivers of tumorigenesis across diverse cancer types.
In this post, we’ll explore the biology of BAF complexes, their role in cancer, and the latest therapeutic insights.

The Role of Chromatin Remodelers
The massive amount of DNA in the human body is truly baffling. Stretched end to end, the DNA from a single somatic cell is about two meters in length, doing the same for all the DNA in the average human would reach the end of the solar system and back!
There are three billion base pairs in our DNA sequences that have to be read, decoded, and translated into proteins to respond to changes to our environment and within our bodies. To organize this process, DNA is compacted into chromatin: DNA wrapped around a core octamer of histone proteins to form nucleosomes, which may then be further compacted into higher-order chromatin fibers. This packaging is a key component of epigenetics, the set of heritable phenotypic changes in gene expression that do not involve changes to the underlying DNA sequence. Once compacted, the tightly wound chromatin can no longer be transcribed unless opened up into euchromatin by various protein complexes and histone modifications. This is where ATP-dependent chromatin remodelers come into play.
ATP-dependent remodelers rely on ATP hydrolysis to displace nucleosomes, allowing access for transcription factors. While multiple protein complexes perform this function, recent studies have shown that there is a particularly high incidence of mutations in cancer in the BAF family of proteins. Researchers have interrogated BAF complexes for years, but now with the advent of –omic technologies and big data, critical roles of these chromatin remodelers in cancer and disease have just begun to be elucidated.
In fact, it was shown that nearly one in four human cancers contain mutations in BAF complex members.1
BAF Complex Mutations: The Cancer Connection
But why is it that mutations in BAF are so prevalent in tumors? The answer to that question is as complicated as the complex itself. We know that individually, many BAF proteins function as tumor suppressors, and their mutations lead to aggressive cancer types such as rhabdoid tumors (no BAF47 expression) and ovarian clear cell carcinomas (no ARID1A expression).2,3 Rhabdoid tumors specifically have an extremely low mutational burden, with BAF47 being one of the few genes mutated.
In other cases, BAF complex members have been shown to be drivers of cancer. Synovial sarcomas are a rare tumor type driven by the oncogenic fusion of the N-terminus of BAF member SS18 to the C-terminus of the SSX1, SSX2, and SSX4 proteins. When incorporated into BAF, the SS18/SSX oncoprotein evicts wild-type BAF47, and effectively hijacks the complex to different parts of the genome, allowing for the expression of oncogenic factors such as Sox2.4,5 This oncogenic hijacking of the BAF complex has also been demonstrated in prostate cancers containing TMPRSS2-ERG fusions.6

IHC analysis of paraffin-embedded human prostate adenocarcinoma (left) and adenoid cystic carcinoma of the trachea (right) using SS18-SSX (E9X9V) XP® Rabbit mAb #72364 (upper) or SS18 (D6I4Z) Rabbit mAb #21792 (lower). Note the absence of the fusion protein in each carcinoma.
BAF, PBAF & ncBAF Complexs
To truly understand how these various subunits are contributing to oncogenesis, we must understand the nuanced mechanisms of the BAF complex. Until recently, very little was actually known about how the BAF complex forms and even the identity of all of its members. As it turns out, there are over 25 different subunits, many of which are mutually exclusive homologs, that comprise three distinct complexes. Each complex starts forming by adding a BAF60 homolog to a BAF155/170 homo or heterodimer. From there, variable subunits get incorporated to form canonical (BAF), non-canonical (ncBAF), or BAF180/PBRM1-containing BAF (PBAF) complexes. Each complex occupies distinct spaces in the genome, suggesting they all have unique functions.

During transcription, DNA replication and repair, chromatin structure is continually modified to expose specific genetic regions and allow DNA-interacting enzymes access to the DNA. ATP-dependent chromatin remodeling complexes including the BAF complex use the energy of ATP hydrolysis to alter chromatin architecture. Explore the ATP-Dependent Chromatin Remodeling Complex pathway, which includes the ISWI, CHD/mi-2, and INO80 Family members and associated CST products.
In addition to the three major complexes, there are also tissue and cell-type-specific BAF complexes. As neural progenitors progress into post-mitotic neurons, subunits are swapped out to form a neural-specific BAF complex (nBAF).7 nBAF plays a critical role in normal brain function, and mutations in various members can cause development disorders such as Coffin-Siris syndrome.8-10 Embryonic stem cells also have a specific variant of BAF, termed esBAF, which is essential for self-renewal and pluripotency.11,12
Understanding the basic biology of the BAF complexes has led to exciting discoveries in previously untreatable tumor types. The identification of the unique ncBAF member BRD9, for instance, has given rise to studies showing it as a critical vulnerability in rhabdoid tumors and synovial sarcomas.13,14
Indeed, the exploration of synthetic lethality is fast becoming an area of interest in BAF research. Tumors seem to need one functional component of mutually exclusive homologs, including Brg1/BRM and ARID1A/B.15,16 In BAF mutant tumors, there is also reliance on other non-BAF related proteins such as PRC2 member Ezh2.17 With wide availability of Ezh2 and BET inhibitors, finding combinational therapies to combat BAF mutant cancer seems to be rife with opportunity.
The Future of BAF Research and Cancer Therapy
While the massive amount of DNA in your cells is indeed BAFfling, the recent flurry of research has given some context to how our cells manage it, while also advancing our understanding of disease states and therapeutic strategies.
Download CST pathways related to epigenetics and chromatin remodeling:

References:
- 23644491
- 9671307
- 20826764
- 23540691
- 29861296
- 30078722
- 17640523
- 28824374
- 22426308
- 22426309
- 19279218
- 19279220
- 30397315
- 31015438
- 24520176
- 24562383
- 26552009