We’re proud to partner with The Michael J. Fox Foundation for Parkinson’s Research (MJFF) to move Parkinson's Disease (PD) research forward. Learn more about the partnership and explore PD resources.
—
Our cells employ highly complex quality control systems to ensure cellular health persists through internal and external stresses. This includes multiple intricate and highly regulated pathways to clear debris, eliminate misfolded proteins, and manage toxic cellular components. Many common neurological disorders, including Parkinson’s disease (PD), can be broadly identified and defined by disruptions to various mechanisms within these complex quality control pathways, which can break down as we age.
Specifically, PD is characterized by loss of function in dopaminergic neurons, leading to a decrease in the neurotransmitter dopamine, which is responsible for coordinating movement. In both sporadic and genetic PD, this loss of function is believed to be caused, in part, by the disruption of various proteins involved in autophagy and lysosomal processing, which lead to the accumulation of misfolded proteins and, ultimately, the death of dopamine neurons. This neuronal loss causes the characteristic tremors seen in PD patients.

The dysfunction and degradation of some of the key processes for maintaining cellular health and homeostasis, such as autophagy, mitophagy, and endolysosomal trafficking, have become an increasing focus for therapeutic intervention in recent years. Developing a better understanding of the mechanisms that contribute to PD—both in its genetic and sporadic forms—is helping pave the way for novel therapeutics.
This blog offers an overview of autophagy, mitophagy, and the endolysosomal system in maintaining neuronal homeostasis. It also examines how disruptions in these pathways contribute to Parkinson's disease. Additionally, it summarizes current techniques for studying lysosomal dysfunction in Parkinson's, referencing key examples from recent scientific literature.
<< Jump to the section Methods for Studying Lysosomal Dysfunction >>
Autophagy, Mitophagy & the Endolysosomal System in Neuronal Homeostasis
To understand the underlying mechanisms and progression of PD, it is essential to recognize the role of autophagy, mitophagy, and the endolysosomal pathway in healthy neurons. As post-mitotic, secretory cells, maintaining neuronal homeostasis is especially important, a process that is made more critical due to the long distance between a neuron’s axon and soma.
.png?width=776&height=337&name=23-EMG-00600-Lysosomal%20%26%20Mitochondrial%20Dysfuntion%20in%20PD%20Blog_Fig%201%20(1).png) When functioning correctly, the Endolysosomal Pathway and Autolysosomal Pathway remove misfolded proteins and damaged organelles from the cell.
When functioning correctly, the Endolysosomal Pathway and Autolysosomal Pathway remove misfolded proteins and damaged organelles from the cell.
Autophagy and endolysosomal trafficking ensure the efficient removal of misfolded, aggregation-prone proteins and damaged organelles, such as dysfunctional mitochondria, thus enabling the maintenance of proper cellular homeostasis.
Cellular Basics: Autophagy
One of the primary cellular degradative pathways, autophagy is an evolutionarily conserved catabolic process that eliminates damaged organelles and misfolded proteins through the selective or non-selective engulfment of cytoplasmic materials in double-membrane autophagosomes. In neurons, autophagosome biogenesis is exclusively initiated in distal axons and synapses1 in order to help replenish the constant need for new proteins and organelles in the periphery.
Autophagy in Neurons: Maintaining the balance between recycled neurotransmitters and autophagy is an activity-dependent control mechanism. In developing neurons, autophagy is required for the pruning of dendritic spines, while in mature neurons, the process is important for cytosolic and pre-synaptic protein sorting and turnover, especially for synaptic vesicles.
Cellular Basics: Mitophagy
Mitophagy is the selective degradation of damaged or excess mitochondria by autophagy, which is important for the maintenance of a healthy mitochondrial network. In neurons, mitophagy can occur in both the cell body and distal axons.
Neuronal Mitophagy: In neurons, under stress conditions, the PINK1 protein and E3 ubiquitin ligase Parkin target defective mitochondria for ubiquitination. This results in the recruitment of autophagic machinery and ubiquitin-binding autophagy receptors, such as p62/SQSTM1 and optineurin. However, it is worth noting that loss of PINK1 or Parkin has been shown to have no effect on basal mitophagy in vivo2,3, suggesting the existence of other PINK1/Parkin-independent mechanisms that cooperate in the clearance of dysfunctional mitochondria.
Cellular Basics: The Endolysosomal Pathway
Present in all cell types, the endolysosomal pathway is a dynamic series of organelles for sorting, modulating, and recycling various membrane cargo brought inside a cell. During the process, endocytic vesicles coated with AP2 internalize cargo from the plasma membrane, which is then either returned to the plasma membrane via recycling endosomes (markers: Rab35, Rab11) or moved along the degradative pathway. Once the AP2 coating is removed, endocytic vesicles containing molecules targeted for lysosomal degradation fuse with early endosomes (markers: Rab5, EEA1), which then mature into late endosomes (marker: Rab7). In the neuronal endolysosomal pathway, late endosomes fuse with autophagosomes (markers: ATG8, LC3) at synaptic terminals. Using their dynein motors, they gradually move along microtubules, acidify, and fuse with lysosomes in the soma to degrade their cargo.
Endolysosomal Trafficking in Neurons: In the post-synapse, the endolysosomal pathway is involved in the recycling and/or degradation of the transmembrane receptors AMPARs and GABAa, depending on synaptic activity. During this process, lysosomes are trafficked in an activity-dependent manner and can be recruited to dendritic spines upon synaptic activation, where they degrade AMPARs and GABAa. This dynamic process is integral to enabling and maintaining neuroplasticity and neurotransmitter release.
Parkinson’s Disease Targets: PARK Genes
The majority of PD cases are sporadic, with less than 10% having a genetic component. However, the genes involved in the inheritable form, termed PARK genes, have been crucial to elucidating its biology. Many PD risk genes are important for maintaining homeostasis and play a role in the endolysosomal pathway, including genes that encode proteins for α-synuclein, LRRK2, VPS35, parkin, PINK1, and DJ1.4
CST offers antibodies to study the functional proteins resulting from PD risk genes across autophagy and the endolysosomal pathway.
- Genes involved in autophagy and the endolysosomal pathway that are PD risk factors:
- Lysosomal enzyme Cathepsin B
- Lysosomal enzyme Glucocerebrosidase (GCase/GBA)
- Vacuolar ATPase subunit ATP6V0A1
- Lysosomal K+ channel TMEM175
- Genes associated with familial PD involved in autophagy and the endolysosomal pathway:
- Synaptojanin-1
- LRRK2
- Lysosomal ATPase ATP13A2 (PARK9)
- PINK1
- Parkin
Lysosomal Disruption & Autophagy in Parkinson's: An α-Syn Example
α-synuclein (α-syn) is one of the most abundant proteins in the brain and its expression suggests a role in plasticity.5 When functioning correctly in healthy neurons, α-syn is a presynaptic protein believed to play a role in neurotransmitter release. The protein is extensively modified post-translation and, as a natively unstructured protein, is thought to adopt many distinct conformations on the basis of the cellular milieu in which it is present.6
The accumulation of misfolded α-syn, and its aggregation into protein inclusions known as Lewy bodies, is believed to be a step in the development and progression of Parkinson's. Under pathological circumstances, α-syn forms aggregates via the assembly of soluble oligomeric intermediates that mature into the insoluble amyloid fibrils found in Lewy bodies. Oligomeric α-syn (o-α-syn) has received considerable attention as a putatively toxic species. Once formed, α-syn aggregates can spread throughout the central nervous system via cell-to-cell propagation, possibly in a prion-like manner.7,8,9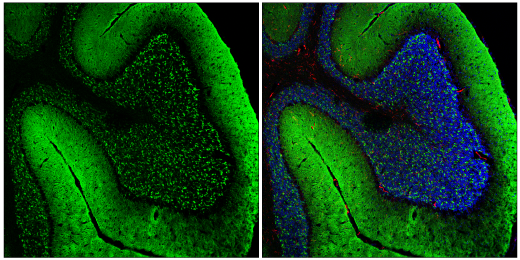
Confocal immunofluorescent analysis of fixed frozen mouse cerebellum labeled with α-Synuclein (E4U2F) XP® Rabbit mAb #51510 (left, green) and co-labeled with F4/80 (BM8.1) Rat mAb #71299 (right, red) and DAPI #4083 (right, blue).
Usually, α-syn is cleared by autophagy, but its aggregates can directly alter the autophagic-lysosomal pathway by reducing ATG7 and increasing mTOR levels, leading to a deficiency in autophagy initiation. While aggregated α-syn is likely to enhance autophagosome formation in both genetic and sporadic PD to meet the higher demand for its degradation,10 its overexpression has also been shown to inhibit autophagosome biogenesis.11 Dopamine-modified α-syn has been shown to further exacerbate this process, which may explain the selective vulnerability of dopaminergic neurons in PD.12
How does α-synuclein avoid degradation in Parkinson's?
- In one study, the α-syn mutants A53T and A30P were able to block their own uptake and the uptake of other substrates by lysosomes for degradation through the chaperone-mediated autophagy pathway in vitro.13
- α-syn aggregates can also disrupt the retrograde transport of AVs, thus impairing autophagosome maturation and fusion with lysosomes.14,15
In PD patients, modified forms of α-syn, such as o-α-syn and phosphorylated α-syn at S129 (pS129α-syn), were significantly increased, with the o-α-syn/t-α-syn ratio being one of the most sensitive (89.3%) and specific (90.6%) tests distinguishing PD from controls.16,17,18,19,20
The Michael J. Fox Foundation has assembled a consortium that is working toward developing a PET tracer that has high affinity and specificity for the aggregated form of α-syn. It remains to be determined whether α-syn will be able to be visualized in a manner similar to amyloid PET imaging.
Methods for Studying Lysosomal Dysfunction
Because of their important role in autophagy and mitophagy, understanding the morphology, exocytosis, acidification, positioning, motility, and function of lysosomes in PD patients is important for further elucidating disease mechanisms. There are various ways to study lysosomal biology using antibody technology, each of which has its own set of pros and cons.
Here, we summarize a few of those methods and suggest a scientific paper you might find useful for further understanding the methods.
-
Live cell LysoTracker: This method provides a useful readout of the number of lysosomes, as well as their size and distribution, in live cells, and is especially useful for therapeutic compound screening. However, this method cannot measure lysosome pH. The fluorescence exhibited by probes is largely independent of pH and accumulates indiscriminately in acidic intracellular organelles. LysoTracker probes label lysosomes as well as late endosomes, and for this reason, the fluorescence signal should not be used as a measure of lysosome pH.
-
Lysosomal Expansion Protocol: Methyl-group-modified amino acid analogs (LEU-ME) can localize to lysosomes, causing an intraluminal osmotic effect that triggers rapid expansion of the lysosomal compartment. After fixation, this technique preserves lysosomal morphology and can be used for the accurate quantification and analysis of lysosomal dimensions. However, this protocol is applicable only for short time-course experiments.
-
Fixed Cell LAMP Staining Protocol: Accessible to any lab with a laser microscope, visualization of lysosomes by fluorescence microscopy is a fast and reliable way to infer possible pathological alterations in lysosomes by studying the distribution and size. Since LAMP1 is present in both late endosomes and lysosomes, a second antibody marker might be needed to specifically identify lysosomes.
-
Lyso-IP Approach Using HA-Tagged TMEM192: This method of lysosomal purification is very fast (10 minutes to isolate pure and intact lysosomes) and uses buffers that are compatible with liquid chromatography and mass spectrometry (LC/MS). However, the overall recovery of lysosomes is low, and the method is only applicable for cells stably expressing HA-TMEM192.
-
Proximity Ligation Assay (PLA Technique): This technique can be used to characterize the over 40 ATG proteins involved in the initiation, elongation, and maturation of the autophagosome. The technique is very versatile for any two proteins in close proximity and can answer many questions in a quantitative manner. It can also be adapted to study protein interactions in paraffin-included tissue samples or in vivo biological tissues. However, results are highly dependent on the quality of the antibody used in the probe, and variation in results is possible due to batch-specific antibody performance. Background signal due to non-specific ligation of oligonucleotides is also possible. These variables can be minimized with the use of highly sensitive and specific antibodies.
In addition to the methods and papers presented above, this resource shares ways to analyze lysosome morphology, positioning, motility, and function and offers an extensive review of current methods used to study lysosomes.
CST Antibody Kits for Studying Parkinson's
CST offers curated antibody sampler kits useful for characterizing mitophagy and the endolysosomal pathway in PD. All CST antibodies are validated using our Hallmarks of Antibody Validation, six complementary strategies that can determine the functionality, specificity, and sensitivity of an antibody in any given assay.
Check out the kits below to see how CST can help move your PD research forward:
- Mitophagy Antibody Sampler Kit #43110
- Autophagy Antibody Sampler Kit #4445
- Parkinson's Research Antibody Sampler Kit #8648
Select References
- Maday S, Holzbaur EL. Autophagosome biogenesis in primary neurons follows an ordered and spatially regulated pathway. Dev Cell. 2014;30(1):71-85. doi:10.1016/j.devcel.2014.06.001
- Lee JJ, Wedow R, Okbay A, et al. Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat Genet. 2018;50(8):1112-1121. Published 2018 Jul 23. doi:10.1038/s41588-018-0147-3
- McWilliams TG, Prescott AR, Montava-Garriga L, et al. Basal Mitophagy Occurs Independently of PINK1 in Mouse Tissues of High Metabolic Demand. Cell Metab. 2018;27(2):439-449.e5. doi:10.1016/j.cmet.2017.12.008
- Panicker N, Ge P, Dawson VL, Dawson TM. The cell biology of Parkinson's disease. J Cell Biol. 2021;220(4):e202012095. doi:10.1083/jcb.202012095
- Sulzer D, Edwards RH. The physiological role of α-synuclein and its relationship to Parkinson's Disease. J Neurochem. 2019;150(5):475-486. doi:10.1111/jnc.14810
- Fayyad M, Salim S, Majbour N, et al. Parkinson's disease biomarkers based on α-synuclein. J Neurochem. 2019;150(5):626-636. doi:10.1111/jnc.14809
- Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson's disease. Nat Med. 2008;14(5):504-506. doi:10.1038/nm1747
- Masuda-Suzukake M, Nonaka T, Hosokawa M, et al. Prion-like spreading of pathological α-synuclein in brain. Brain. 2013;136(Pt 4):1128-1138. doi:10.1093/brain/awt037
- Recasens A, Dehay B, Bové J, et al. Lewy body extracts from Parkinson disease brains trigger α-synuclein pathology and neurodegeneration in mice and monkeys. Ann Neurol. 2014;75(3):351-362. doi:10.1002/ana.24066
- Wong YC, Holzbaur EL. Temporal dynamics of PARK2/parkin and OPTN/optineurin recruitment during the mitophagy of damaged mitochondria. Autophagy. 2015;11(2):422-424. doi:10.1080/15548627.2015.1009792
- Winslow AR, Chen CW, Corrochano S, et al. α-Synuclein impairs macroautophagy: implications for Parkinson's disease. J Cell Biol. 2010;190(6):1023-1037. doi:10.1083/jcb.201003122
- Martinez-Vicente M, Talloczy Z, Kaushik S, et al. Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J Clin Invest. 2008;118(2):777-788. doi:10.1172/JCI32806
- Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305(5688):1292-1295. doi:10.1126/science.1101738
- Tanik SA, Schultheiss CE, Volpicelli-Daley LA, Brunden KR, Lee VM. Lewy body-like α-synuclein aggregates resist degradation and impair macroautophagy. J Biol Chem. 2013;288(21):15194-15210. doi:10.1074/jbc.M113.457408
- Volpicelli-Daley LA, Luk KC, Lee VM. Addition of exogenous α-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous α-synuclein to Lewy body and Lewy neurite-like aggregates. Nat Protoc. 2014;9(9):2135-2146. doi:10.1038/nprot.2014.143
- Parnetti L, Chiasserini D, Persichetti E, et al. Cerebrospinal fluid lysosomal enzymes and alpha-synuclein in Parkinson's disease. Mov Disord. 2014;29(8):1019-1027. doi:10.1002/mds.25772
- Parnetti L, Farotti L, Eusebi P, et al. Differential role of CSF alpha-synuclein species, tau, and Aβ42 in Parkinson's Disease. Front Aging Neurosci. 2014;6:53. Published 2014 Mar 31. doi:10.3389/fnagi.2014.00053
- Tokuda T, Salem SA, Allsop D, et al. Decreased alpha-synuclein in cerebrospinal fluid of aged individuals and subjects with Parkinson's disease. Biochem Biophys Res Commun. 2006;349(1):162-166. doi:10.1016/j.bbrc.2006.08.024
- Hansson O, Hall S, Ohrfelt A, et al. Levels of cerebrospinal fluid α-synuclein oligomers are increased in Parkinson's disease with dementia and dementia with Lewy bodies compared to Alzheimer's disease. Alzheimer's Res Ther. 2014;6(3):25. Published 2014 May 7. doi:10.1186/alzrt255
- Aasly JO, Johansen KK, Brønstad G, et al. Elevated levels of cerebrospinal fluid α-synuclein oligomers in healthy asymptomatic LRRK2 mutation carriers. Front Aging Neurosci. 2014;6:248. Published 2014 Sep 25. doi:10.3389/fnagi.2014.00248